I have recently been doing a course (along with a bunch of postgraduate students) on Massively Parallel Sequencing, also known as Next Generation Sequencing (NGS). This was a partially successful attempt to teach an old dog some new tricks. More to the point, it has prompted me to think about NGS in relation to phylogenetic networks. Most of the published discussions have focussed on trees, rather than networks.

NGS can potentially provide a fast and cost-effective means of generating multilocus sequence data for phylogenetics (Rannala & Yang 2008; McCormack et al. 2013; Moriarty Lemmon & Lemmon 2013). Unfortunately, the cost for the number of samples typically employed in phylogenetics is currently still beyond the reach of most researchers.
NGS and phylogenetics
Nevertheless, we are sometimes told things like: "The fields of phylogenetics and phylogeography are on the cusp of a revolution, enabled by the rapid expansion of genomic resources and explosion of new genome sequencing technologies." This is probably over-stating the case, as noted by McCormack et al. (2013):
Despite this obvious potential, NGS has been slow to take root in phylogeography and phylogenetics compared to other fields like metagenomics and disease genetics. We suggest that this lag has been caused by four specific aspects of phylogeographic and phylogenetic research: the predominant focus on non-model organisms, the need for sequencing large numbers of samples per species, the lack of consensus regarding library preparation protocols for particular research questions, and the transitional state of the technology (whole-genome data are still neither cost-effective, nor even desirable for phylogeography and phylogenetics, but are paradoxically easier to collect).
Another issue is the historical importance of utilizing gene trees in phylogeography and phylogenetics. Gene trees are most robustly inferred from loci with high information content, for example, a non-recombining locus containing a series of linked SNPs. Individual SNPs, on the other hand, have low information content on a per-locus basis and have been used predominately with classification methods such as Structure and Principal components analysis ... While distance-based genealogies and phylogenies can be built from unlinked SNPs, this ignores models of molecular substitution and probabilistic tree-searching algorithms that have led to more robust phylogenetic inference in the last several decades.Furthermore, no-one has yet shown that many of the questions currently being asked by phylogeneticists will actually benefit from genomic data. We may well be able to answer some new questions, but that is quite a different thing from a revolution. The essence here is that in science the questions must come first. Collecting data for the sake of it is usually unproductive. So, we need a clear demonstration that genomics is actually needed in phylogenetics (as opposed to other disciplines, where it may indeed be very useful). If increased volume of data will solve a phylogenetic problem then that is good, but there is no necessary reason to expect that it will happen. Statistically, the extra data can lead to improved precision but not necessarily improved accuracy. In science, targeted data collection has always been the most productive approach to any clearly stated experimental question.
For example, the estimated relationships among humans, chimpanzees, and gorillas did not change as a result of genome sampling (Galtier and Daubin 2008), nor did those of malaria species (Kuo et al. 2008), nor those of mammal superorders (Hallström and Janke 2010). (I have discussed the mammal example in a previous blog post: Why are there conflicting placental roots?). In all three cases, the relationships were just as complex after the genome sequencing as before — the resolution of controversial branches in our trees did not occur as a result of increased access to character data.
In this sense, a small sample of representative gene sequences should reveal just as much of the genealogical truth as will a genome-wide sample. A more recent empirical example is presented by O'Neill et al. (2013), who found that including less informative loci added so much noise to the phylogenetic signal that the analysis eventually broke down. The issue here is that as data volume increases so does the potential occurrence of systematic bias due to model mis-specification.
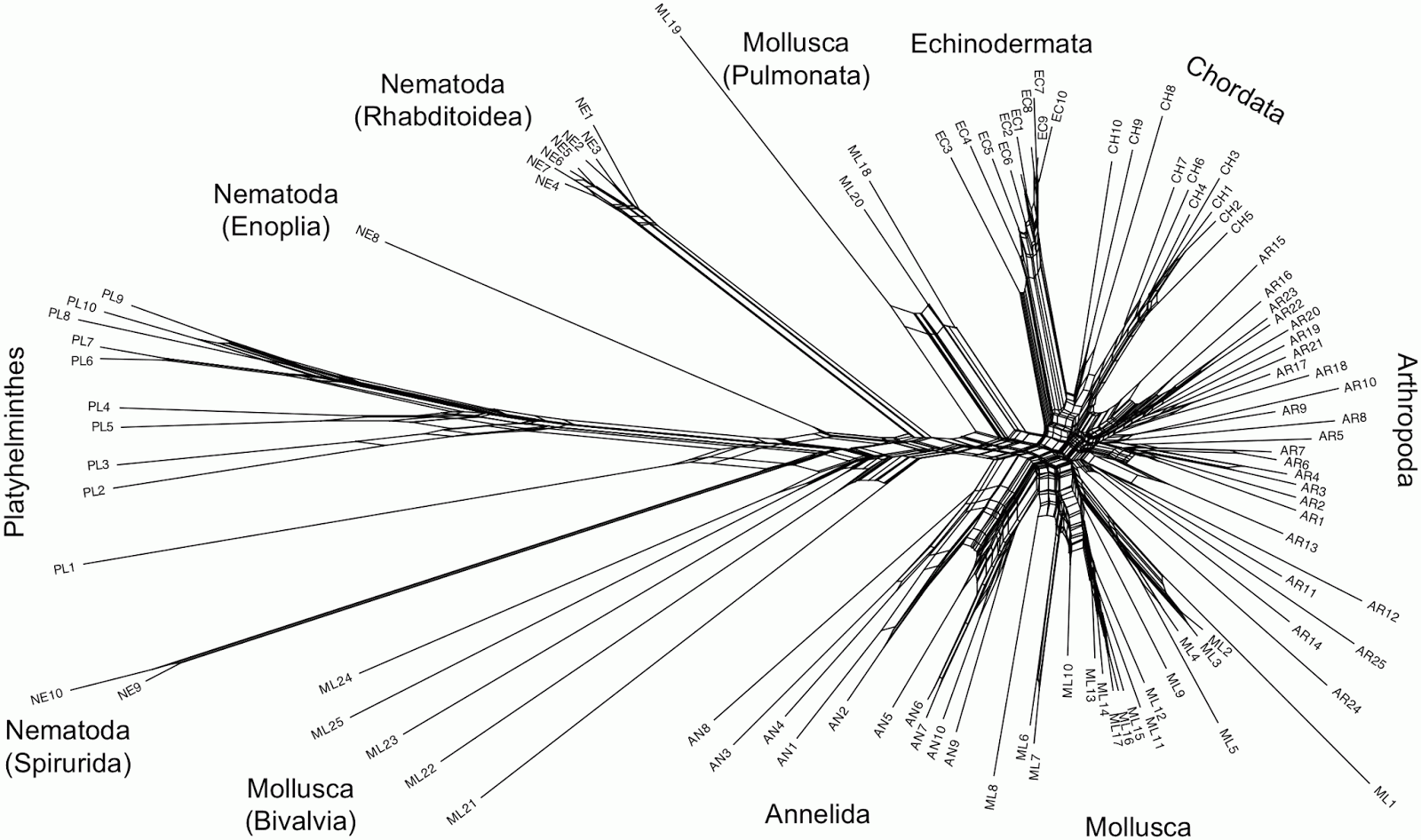
This sort of problem can easily be visualized using phylogenetic networks, in which genome-scale data frequently produce unresolved bushes rather than tree-like phylogenies. I have provided a couple of examples in a previous post (When is there support for a large phylogeny?). Another example is provided by Beiko (2011), which I have reproduced below.
This all suggests that we will need to think carefully about how to apply phylogenetic networks to genome-scale data. Much of the lack of resolution may very well come from the nature of NGS, rather than from the actual evolutionary history.

NGS and networks
There are a number of potential problems with NGS. These may not matter so much for tree-building algorithms, but it is a different matter for networks.
[1] Increased homoplasy due to sequencing errors
An error rate of even 0.01% is considered good in NGS (eg. Roche 454: 1%; Illumina HiSeq: 0.1%; Life SOLiD: 0.01%), but when this is extrapolated to the genome scale it results in thousands of errors. Networks are sensitive to this magnitude of stochastic error. Indeed, I have already written about the use of phylogenetic networks specifically to identify data errors (Checking data errors with phylogenetic networks).
[2] Increased homoplasy due to intra-gene processes
These include substitutions, deletions, duplications (especially tandem repeats), inversions, and translocations. These processes can potentially reveal evolutionary history, but we have little idea about how best to process the data in a way that will reveal that history. Currently, we deal with this by lumping most of the processes together as "indels".
[3] Increased homoplasy due to inter-gene processes
The most common processes known to confound attempts to identify reticulate evolution are incomplete lineage sorting and gene duplication–loss. There are several methods available for addressing these issues in the context of estimating phylogenetic trees, but their applicability to networks is still being assessed.
[4] Increased homoplasy in non-coding regions
Sanger-style sequencing is usually targeted towards gene-coding regions or their introns, but genome-scale data can include what is currently called "junk DNA". The evolutionary processes in these regions are unknown, as is their applicability to phylogenetic analysis.
[5] Inadequacies due to data-processing methods
The analysis of NGS data is often a black art — each paper seems to provide its own way of processing the data. This has been a cause of concern expressed in the literature (e.g. Check Hayden 2012; Editorial 2012a, 2012b; MacArthur 2012), especially in the light of the poor documentation and archiving of bioinformatics programs. I have discussed this issue in some previous posts (Poor bioinformatics?; Archiving of bioinformatics software). Perhaps the most talked-about problem is ascertainment bias — there is a brief discussion of this at the end of this post.
Network analysis of NGS data
All of this might make the application of networks to phylogenomics problematic in many cases, because we already have enough challenges dealing with the data from Sanger-style sequencing, without having them be orders of magnitude worse. It will therefore be very interesting to see what emerges from the current attempts to apply phylogenetic networks to NGS data.
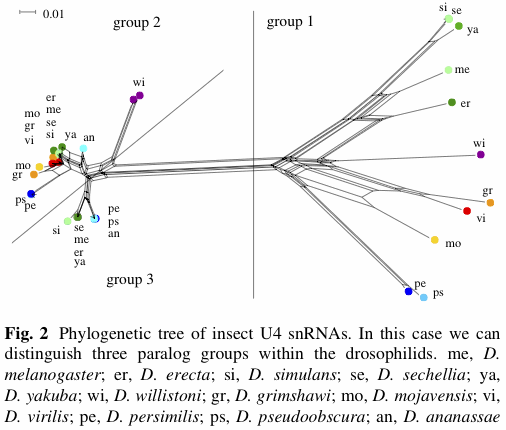
There have been a few applications of EDA (exploratory data analysis) programs such as SplitsTree, mostly involving bacteria and viruses, and often in the context of detecting recombination. Not all of these studies have produced networks that look bushy, as shown by the example below (from Söderlund et al. 2013). SplitsTree is mostly limited by the number of samples not by the number of characters, so that genomic data are not a particular analysis issue for algorithms such as neighbor-net. However, you might like to calculate your inter-sample distances outside the program, unless you want the simple p-distance. (Popular genome-scale alternatives include Fst.)
There have also been programs developed for the study of admixture (a.k.a. introgression) in human genomes, such as TreeMix, AdmixTools, and MixMapper, and these might repay wider exploration. I have discussed some of these programs in a previous post (Admixture graphs – evolutionary networks for population biology). Essentially, they first construct a tree and then add reticulations based on various criteria. As is usual with this approach, there is the problem of constructing the initial tree in the presence of reticulation processes, and there seems to be no clear criterion about when to stop adding reticulations — optimization criteria always increase as reticulations are added, so that increasingly complex networks will always be preferred mathematically.

Note — a common data-processing problem
The following explanation of one type of ascertainment bias is adapted from the Fluxus Engineering web site:
For each DNA sample, a large number of short sequences are generated by the NGS sampling. Genomic variants are estimated from the consensus of these NGS sequences, after filtering the sequences for artifacts. Variant lists are never complete — the greater the sequence length, the greater the fraction of the genome that can be sequenced, but there are always uncharted regions which vary from sample to sample. The sampled genome sequences are then compared to a reference genome. NGS software usually reports SNP variants only if they do not match the reference genotype, and if there is sufficient evidence that they are non-reference. Non-reported variants do not necessarily match the reference genotype — they can just as well be sequencing failures, or coverage gaps, or insufficient evidence for a non-reference variant. Networks generated from such data are likely to consist largely of artifacts.References
Beiko RG (2011) Telling the whole story in a 10,000-genome world. Biology Direct 6: 34.
Check Hayden E (2012) RNA studies under fire. Nature 484: 428.
Editorial (2012a) Must try harder. Nature 483: 509.
Editorial (2012b) Error prone. Nature 487: 406.
Galtier N, Daubin V (2008) Dealing with incongruence in phylogenomic analyses. Philosophical Transactions of the Royal Society of London, Series B: Biological Sciences 363: 4023-4029.
Hallström BM, Janke A (2010) Mammalian evolution may not be strictly bifurcating. Molecular Biology & Evolution 27: 2804-2816.
Kuo C-H, Wares JP, Kissinger JC (2008) The Apicomplexan whole-genome phylogeny: an analysis of incongruence among gene trees. Molecular Biology & Evolution 25: 2689-2698.
Moriarty Lemmon E, Lemmon AR (2013) High-throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution & Systematics 2013. 44: 19.1–19.23.
MacArthur D (2012) Face up to false positives. Nature 487: 427-428.
McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT (2013) Applications of next-generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution 66: 526-538.
O'Neill EM, Schwartz R, Bullock CT, Williams JS, Shaffer HB, Aguilar-Miguel X, Parra-Olea G, Weisrock DW (2013) Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex. Molecular Ecology 22: 111-129.
Rannala B, Yang Z (2008) Phylogenetic inference using whole genomes. Annual Review of Genomics and Human Genetics 9: 217-231.
Söderlund R, Jernberg C, Källman C, Hedenström I, Eriksson E, Bongcam-Rudloff E, Aspán A (2013) Rapid whole genome sequencing investigation of a familial outbreak of E. coli O121:H19 with a sheep farm as the suspected source. EMBnet Journal 19 suppl.A: 89-90.