The procedures by which linguists sample data when carrying out phylogenetic analyses of languages are sometimes fundamentally different from the methods applied in biology. This is particularly obvious in the matter of the sampling of data for analysis, which I will discuss in this post.
Sampling data in historical linguistics
The reason for the difference is straightforward: while biologists can now sample whole genomes and search across those genomes for shared word families, linguists cannot sample the whole lexicon of several languages. The problem is not that we could not apply cognate detection methods to whole dictionaries. In fact there are recent attempts that try to do exactly this (Arnaud et al. 2017). The problem is that we simply do not know exactly how many words we can find in any given language.
For example, the Duden, a large lexicon of the German language, for example, recently added 5000 more words, mostly due to recent technological innovations, which then lead to new words which we frequently use in German, such as twittern "to tweet", Tablet "tablet computer", or Drohnenangriff "drone attack". In total, it now lists 145,000 words, and the majority of these words has been coined in complex processes involving language-internal derivation of new word forms, but also by a large amount of borrowing, as one can see from the three examples.
One could argue that we should only sample those words which most of the speakers in a given language know, but even there we are far from being able to provide reliable statistics, not to speak of the fact that it is also possible that these numbers vary greatly across different language families and cultural and sociolinguistic backgrounds. Brysbaert et al. (2016), for example, estimate that
an average 20-year-old native speaker of American English knows 42,000 lemmas and 4,200 non-transparent multiword expressions, derived from 11,100 word families.But in order to count as "near-native" in a certain language, including the ability to pursue studies at a university, the Common European Framework of Reference for Languages, requires only between 4000 and 5000 words (Milton 2010, see also List et al. 2016). How many word families this includes is not clear, and may, again, depend directly on the target language.
Lexicostatistics
When Morris Swadesh (1909-1967) established the discipline of lexicostatistics, which represented the first attempt to approach the problems we face in historical linguistics with the help of quantitative methods. He started from a sample of 215 concepts (Swadesh 1950), which he later reduced to only 100 (Swadesh 1955), because he was afraid that some concepts would often be denoted by words that are borrowed, or that would simply not be expressed by single words in certain language families. Since then, linguists have been trying to refine this list further, either by modifying it (Starostin 1991 added 10 more concepts to Swadesh's list of 100 concepts), or by reducing it even further (Holman et al. 2008 reduced the list to 40 concepts).
While it is not essential how many concepts we use in the end, it is important to understand that we do not start simply by comparing words in our current phylogenetic approaches, but instead we sample parts of the lexicon of our languages with the help of a list of comparative concepts (Haspelmath 2010), which we then consecutively translate into the target languages. This sampling procedure was not necessarily invented by Morris Swadesh, but he was first to establish its broader use, and we have directly inherited this procedure of sampling when applying our phylogenetic methods (see this earlier post for details on lexicostatistics).
Synonymy in linguistic datasets
Having inherited the procedure, we have also inherited its problems, and, unfortunately, there are many problems involved with this sampling procedure. Not only do we have difficulties determining a universal diagnostic test list that could be applied to all languages, we also have considerable problems in standardizing the procedure of translating a comparative concept into the target languages, especially when the concepts are only loosely defined. The concept "to kill", for example, seems to be a rather straightforward example at first sight. In German, however, we have two words that could express this meaning equally well: töten (cognate with English dead) and umbringen (partially cognate with English to bring). In fact, as with all languages in the world, there are many more words for "to kill" in German, but these can easily be filtered out, as they usually are euphemisms, such as eliminieren "to eliminate", or neutralisieren "to neutralize". The words töten and umbringen, however, are extremely difficult to distinguish with respect to their meaning, and speakers often use them interchangeably, depending, perhaps, on register (töten being more formal). But even for me as a native speaker of German, it is incredibly difficult to tell when I use which word.
One solution to making a decision as to which of the words is more basic could be corpus studies. By counting how often and in which situations one term is used in a large corpus of German speech, we might be able to determine which of the two words comes closer to the concept "to kill" (see Starostin 2013 for a very elegant example for the problem of words for "dog" in Chinese). But in most cases where we compile lists of languages, we do not have the necessary corpora.
Furthermore, since corpus studies on competing forms for a given concept are extremely rare in linguistics, we cannot exclude the possibility that the frequency of two words expressing the same concept is in the end the same, and the words just represent a state of equilibrium in which speakers use them interchangeably. Whether we like it or not, we have to accept that there is no general principle to avoid these cases of synymony when compiling our datasets for phylogenetic analyses.
Tossing coins
What should linguists do in such a situation, when they are about to compile the dataset that they want to analyze with the modern phylogenetic methods, in order to reconstruct some eye-catching phylogenetic trees? In the early days of lexicostatistics, scholars recommended being very strict, demanding that only one word in a given language should represent one comparative concept. In cases like German töten and umbringen, they recommended to toss a coin (Gudschinsky 1956), in order to guarantee that the procedure was as objective as possible.
Later on, scholars relaxed the criteria, and just accepted that in a few — hopefully very few — cases there would be more than one word representing a comparative concept in a given language. This principle has not changed with the quantitative turn in historical linguistics. In fact, thanks to the procedure by which cognate sets across concept slots are dichotomized in a second step, scholars who only care for the phylogenetic analyses and not for the real data may easily overlook that the Nexus file from which they try to infer the ancestry of a given language family may list a large amount of synonyms, where the classical scholars simply did not know how to translate one of their diagnostic concepts into the target languages.
Testing the impact of synonymy on phylogenetic reconstruction
The obvious question to ask at this stage is: does this actually matter? Can't we just ignore it and trust that our phylogenetic approaches are sophisticated enough to find the major signals in the data, so that we can just ignore the problem of synonymy in linguistic datasets? In an early study, almost 10 years ago, when I was still a greenhorn in computing, I made an initial study of the problem of extensive synonymy, but it never made it into a publication, since we had to shorten our more general study, of which the synonymy test was only a small part. This study has been online since 2010 (Geisler and List 2010), but is still awaiting publication; and instead of including my quantitative test on the impact of extensive synonymy on phylogenetic reconstruction, we just mentioned the problem briefly.
Given that the problem of extensive synonymy turned up frequently in recent discussions with colleagues working on phylogenetic reconstruction in linguistics, I decided that I should finally close this chapter of my life, and resume the analyses that had been sleeping in my computer for the last 10 years.
The approach is very straightforward. If we want to test whether the choice of translations leaves traces in phylogenetic analyses, we can just take the pioneers of lexicostatistics literally, and conduct a series of coin-tossing experiments. We start from a "normal" dataset that people use in phylogenetic studies. These datasets usually contain a certain amount of synonymy (not extremely many, but it is not surprising to find two, three, or even four translations in the datasets that have been analysed in the recent years). If we now have the computer toss a coin in each situation where only one word should be chosen, we can easily create a large sample of datasets each of which is synonym free. Analysing these datasets and comparing the resulting trees is again straightforward.
I wrote some Python code, based on our LingPy library for computational tasks in historical linguistics (List et al. 2017), and selected four datasets, which are publicly available, for my studies, namely: one Indo-European dataset (Dunn 2012), one Pama-Nyungan dataset (Australian languages, Bowern and Atkinson 2012), one Austronesian dataset (Greenhill et al. 2008), and one Austro-Asiatic dataset (Sidwell 2015). The following table lists some basic information about the number of concepts, languages, and the average synonymy, i.e., the average number of words that a concept expresses in the data.
| Dataset | Concepts | Languages | Synonymy | |
|---|---|---|---|---|
| Austro-Asiatic | 200 | 58 | 1.08 | |
| Austronesian | 210 | 45 | 1.12 | |
| Indo-European | 208 | 58 | 1.16 | |
| Pama-Nyungan | 183 | 67 | 1.1 |
For each dataset, I made 1000 coin-tossing trials, in which I randomly picked only one word where more than one word would have been given as the translation of a given concept in a given language. I then computed a phylogeny of each newly created dataset with the help of the Neighbor-joining algorithm on the distance matrix of shared cognates (Saitou and Nei 1987). In order to compare the trees, I employed the general Robinson-Foulds distance, as implemented in LingPy by Taraka Rama. Since I did not have time to wait to compare all 1000 trees against each other (as this takes a long time when computing the analyses for four datasets), I randomly sampled 1000 tree pairs. It is, however, easy to repeat the results and compute the distances for all tree pairs exhaustively. The code and the data that I used can be found online at GitHub (github.com/lingpy/toss-a-coin).
Some results
As shown in the following table, where I added the averaged generalized Robinson-Foulds distances for the pairwise tree comparisons, it becomes obvious that — at least for distance-based phylogenetic calculations — the problem of extensive synonymy and choice of translational equivalents has an immediate impact on phylogenetic reconstruction. In fact, the average differences reported here are higher than the ones we find when comparing phylogenetic reconstruction based on automatic pipelines with phylogenetic reconstruction based on manual annotation (Jäger 2013).
| Dataset | Concepts | Languages | Synonymy | Average GRF |
|---|---|---|---|---|
| Austro-Asiatic | 200 | 58 | 1.08 | 0.20 |
| Austronesian | 210 | 45 | 1.12 | 0.19 |
| Indo-European | 208 | 58 | 1.16 | 0.59 |
| Pama-Nyungan | 183 | 67 | 1.1 | 0.22 |
The most impressive example is for the Indo-European dataset, where we have an incredible average distance of 0.59. This result almost seems surreal, and at first I thought that it was my lazy sampling procedure that introduced the bias. But a second trial confirmed the distance (0.62), and when comparing each of the 1000 trial trees with the tree we receive when not excluding the synonyms, the distance
is even slightly higher (0.64).
When looking at the consensus network of the 1000 trees (created with SplitsTree4, Huson et al. 2006), using no threshold (to make sure that the full variation could be traced), and the mean for the calculation of the branch lengths, which is shown below, we can see that the variation introduced by the synonyms is indeed real.
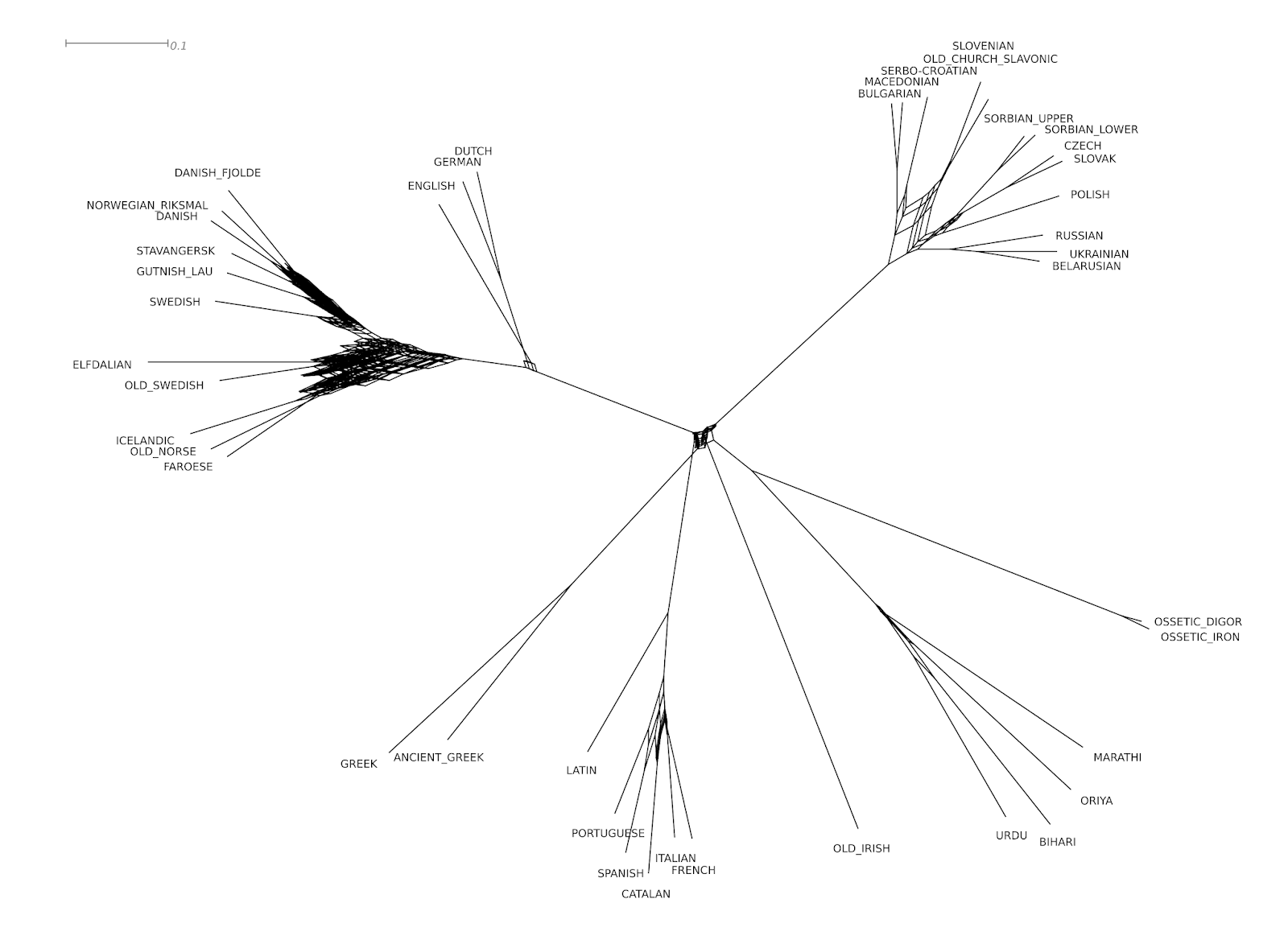 |
| The consensus network of the 1000 tree sample for the Indo-European language sample |
Notably, the Germanic languages are highly incompatible, followed by Slavic and Romance. In addition, we find quite a lot of variation in the root. Furthermore, when looking the at the table below, which shows the ten languages that have the largest number of synonyms in the Indo-European data, we can see that most of them belong to the highly incompatible Germanic branch.
| Language | Subgroup | Synonymous Concepts |
|---|---|---|
| OLD_NORSE | Germanic | 83 |
| FAROESE | Germanic | 77 |
| SWEDISH | Germanic | 68 |
| OLD_SWEDISH | Germanic | 65 |
| ICELANDIC | Germanic | 64 |
| OLD_IRISH | Celtic | 61 |
| NORWEGIAN_RIKSMAL | Germanic | 54 |
| GUTNISH_LAU | Germanic | 50 |
| ORIYA | Indo-Aryan | 50 |
| ANCIENT_GREEK | Greek | 46 |
Conclusion
This study should be taken with some due care, as it is a preliminary experiment, and I have only tested it on four datasets, using a rather rough procedure of sampling the distances. It is perfectly possible that Bayesian methods (as they are "traditionally" used for phylogenetic analyses in historical linguistics now) can deal with this problem much better than distance-based approaches. It is also clear that by sampling the trees in a more rigorous manner (eg. by setting a threshold to include only those splits which occur frequently enough), the network will look much more tree like.
However, even if it turns out that the results are exaggerating the situation due to some theoretical or practical errors in my experiment, I think that we can no longer ignore the impact that our data decisions have on the phylogenies we produce. I hope that this preliminary study can eventually lead to some fruitful discussions in our field that may help us to improve our standards of data annotation.
I should also make it clear that this is in part already happening. Our colleagues from Moscow State University (lead by George Starostin in the form of the Global Lexicostatistical Database project) try very hard to improve the procedure by which translational equivalents are selected for the languages they investigate. The same applies to colleagues from our department in Jena who are working on an ambitious database for the Indo-European languages.
In addition to linguists trying to improve the way they sample their data, however, I hope that our computational experts could also begin to take the problem of data sampling in historical linguistics more seriously. A phylogenetic analysis does not start with a Nexus file. Especially in historical linguistics, where we often have very detailed accounts of individual word histories (derived from our qualitative methods), we need to work harder to integrate software solutions and qualitative studies.
References
Arnaud, A., D. Beck, and G. Kondrak (2017) Identifying cognate sets across dictionaries of related languages. In: Proceedings of the 2017 Conference on Empirical Methods in Natural Language Processing 2509-2518.
Bowern, C. and Q. Atkinson (2012) Computational phylogenetics of the internal structure of Pama-Nguyan. Language 88. 817-845.
Brysbaert, M., M. Stevens, P. Mandera, and E. Keuleers (2016) How many words do we know? Practical estimates of vocabulary size dependent on word definition, the degree of language input and the participant’s age. Frontiers in Psychology 7. 1116.
Dunn, M. (ed.) (2012) Indo-European Lexical Cognacy Database (IELex). http://ielex.mpi.nl/.
Geisler, H. and J.-M. List (2010) Beautiful trees on unstable ground: notes on the data problem in lexicostatistics. In: Hettrich, H. (ed.) Die Ausbreitung des Indogermanischen. Thesen aus Sprachwissenschaft, Archäologie und Genetik. Reichert: Wiesbaden.
Greenhill, S., R. Blust, and R. Gray (2008) The Austronesian Basic Vocabulary Database: From bioinformatics to lexomics. Evolutionary Bioinformatics 4. 271-283.
Gudschinsky, S. (1956) The ABC’s of lexicostatistics (glottochronology). Word 12.2. 175-210.
Haspelmath, M. (2010) Comparative concepts and descriptive categories. Language 86.3. 663-687.
Holman, E., S. Wichmann, C. Brown, V. Velupillai, A. Müller, and D. Bakker (2008) Explorations in automated lexicostatistics. Folia Linguistica 20.3. 116-121.
Huson, D. and D. Bryant (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23.2. 254-267.
Jäger, G. (2013) Phylogenetic inference from word lists using weighted alignment with empirical determined weights. Language Dynamics and Change 3.2. 245-291.
List, J.-M., J. Pathmanathan, P. Lopez, and E. Bapteste (2016) Unity and disunity in evolutionary sciences: process-based analogies open common research avenues for biology and linguistics. Biology Direct 11.39. 1-17.
List, J.-M., S. Greenhill, and R. Forkel (2017) LingPy. A Python Library For Quantitative Tasks in Historical Linguistics. Software Package. Version 2.6. Max Planck Institute for the Science of Human History: Jena.
Milton, J. (2010) The development of vocabulary breadth across the CEFR levels: a common basis for the elaboration of language syllabuses, curriculum guidelines, examinations, and textbooks across Europe. In: Bartning, I., M. Martin, and I. Vedder (eds.) Communicative Proficiency and Linguistic Development: Intersections Between SLA and Language Testing Research. Eurosla: York. 211-232.
Saitou, N. and M. Nei (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4.4. 406-425.
Sidwell, P. (2015) Austroasiatic Dataset for Phylogenetic Analysis: 2015 version. Mon-Khmer Studies (Notes, Reviews, Data-Papers) 44. lxviii-ccclvii.
Starostin, S. (1991) Altajskaja problema i proischo\vzdenije japonskogo jazyka [The Altaic problem and the origin of the Japanese language]. Nauka: Moscow.
Starostin, G. (2013) K probleme dvuch sobak v klassi\cceskom kitajskom jazyke: canis comestibilis vs. canis venaticus? [On the problem of two words for dog in Classical Chinese: edible vs. hunting dog?]. In: Grincer, N., M. Rusanov, L. Kogan, G. Starostin, and N. \cCalisova (eds.) Institutionis conditori: Ilje Sergejevi\ccu Smirnovy.[In honor of Ilja Sergejevi\cc Smirnov].L. RGGU: Moscow. 269-283.
Swadesh, M. (1950) Salish internal relationships. International Journal of American Linguistics 16.4. 157-167.
Swadesh, M. (1955) Towards greater accuracy in lexicostatistic dating. International Journal of American Linguistics 21.2. 121-137.