This is Part 2 of a 2-part blog series.
Part 1 covered some history, while this post has three (more) recently published matrices, and the take-home message.
Jumping forward in time, welcome to the 21st century
In
Part 1, I showed several networks generated based on some early phylogenetic matrices published in the first volumes of the journal
Cladistics. In this post, we will look at the most recent data matrices and trees uploaded to TreeB
ASE, covering the past seven years.
Nearly a generation later, and facing the "molecular revolution", some researchers (fortunately) still compile morphological matrices. This is an often overlooked but important work: genes and genomes can be sequenced by machines, and the only thing we need to do is to feed these machine-generated data into other powerful machines (and programs) to get a phylogenetic tree, or network. But no software and computer cluster can (so far) study anatomy, and generate a morphological matrix. The latter is paramount when we want to put fossils, usually devoid of DNA, in a (molecular) phylogenetic context. We need to do this when we aim to reconstruct histories in space
and time.
Nevertheless, we can't ignore the fact that these important data are (still) far from tree-like. What holds for the matrices of the 80's (see the end of
Part 1), still applies now.
So, let's have a look at the three most recent data sets (one morphological, two molecular) published in
Cladistics that have their data matrix in TreeB
ASE.
The morphological dataset
Beutel et al. (2011; submission S11976) provided a "robust phylogeny of ... Holometabola", and note in their abstract: "Our results show little congruence with studies based on rRNA, but
confirm most clades retrieved in a recent study based on nuclear genes."
Without having read the study, I can guess which clades (likely used here as a synonym for monophyletic group; but see
David's post on Hennig and Cladistics) were confirmed. The data matrix contains: 356 multistate, with up to six states, characters scored
and annotated for 34 taxa, including polymorphisms and some gaps ("–")
viz missing data ("?"). Just by looking at the Neighbor-net inferred from this matrix. (Standard tree- or network-inference doesn't differ between gaps and missing data, but some people find it important to distinguish between "not applicable" and "not known" in a matrix.)
 |
| Neighbor-net inferred from simple pairwise distances computed based on Beutel et al.'s matrix. Brackets show my ad hoc assessment of candidates for monophyla (here: likely represented by clades in no matter how optimized trees). |
How did I postulate the monophyla? By deduction: if two or more OTUs are much more similar to each other than to anything else in the matrix, they likely are part of the same evolutionary lineage, ie. have a common origin (= monophyletic in a pre-Hennigian sense). This, when the matrix well covers the group and morphospace, has a good chance to be inclusive (= monophyletic fide Hennig; for the covered OTUs). This is especially so when there is a good deal of homoplasy — the provided tree has a CI of 0.44 and RC of 0.33: convergences should be more randomly distributed than lineage-specific/-conserved traits. The latter don't need to be (or were, at some point in time) synapomorphies, shared derived unique traits, but could be diagnostic suites of characters that evolved in parallel within a lineage and passed on to all (or most) of the descendants.
The first molecular dataset
Let's look at the signal in the two molecular matrices.
In 2016,
Gaspar and Almeida (submission S19167) tested generic circumscriptions in a group of ferns by "assembl[ing] the broadest dataset thus far, from three plastid regions (rbcL, rps4-trnS, trnL-trnF) ... includ[ing] 158 taxa and 178 newly generated sequences". They found: "three subfamilies each corresponding to a highly supported clade across
all analyses (maximum parsimony, Bayesian inference, and maximum
likelihood)."
The total matrix has 3250 characters, of which 1641 are constant and 1189 are parsimony-informative. This is a quite a lot for such a matrix, and, by itself, rules out parsimony for tree-inference. If half of the nucleotide sites are variable, then the rate of character change was high, and parsimony is statistically only robust, when the rate of change was low. High mutation rates or high level of divergence may also pose problems for distance methods and other optimality criteria, all closely related to parsimony.
The file includes three trees, labelled "vero" (which, in Italian, means "true"), "Fig._1" and "MPT". "Vero" and "Fig._1" come with branch lengths; judging from the values (<< 1), they are probabilistic trees (of some sort); the "MPT" is (as usual) provided as a cladogram without branch-lengths. It may be that the authors had to add the parsimony tree just to fulfill editorial policies, while being convinced "vero" is the much better tree. "Vero" is a fully resolved tree (the ML tree?), while "Fig._1" (Bayesian?) and "MPT" include polytomies.
Using PAUP*'s "describe" function, we learn that the "MPT" is 5101 steps long and has a CI of 0.41 and RC of 0.33. Nucleotide sequence data can be notoriously homoplasious, as we repeat the same four states into infinity and have to deal with an unknown but usually significant amount of back mutations. This adds to the other problems for parsimony:
- transitions are more likely to happen than transversions; and
- in coding gene regions, such as the rbcL, some sites (3rd codon positions) mutate much faster than others.
Still, parsimony trees are not necessarily wrong. Neither are NJ trees; and there are also datasets where probabilistic methods struggle, eg. when the likelihood surface of the treespace is flat.
So, the first question is: how different are the three trees provided? Rather than having to show three graphs, we can show the (strict) Consensus network of those trees.
 |
| A strict consensus network summarizing the topologies of the three trees provided in the TreeBASE submission of |
The main difference is between "vero" and the other two — "Fig. 1" and the "MPT" are very similar (and both include polytomies). There are three main scenarios for a Consensus network like this with respect to the high portion of variable sites:
- "Fig. 1" is a Jukes-Cantor model-based tree,
- "Fig. 1" is an uncorrected p-distance based tree, or
- most of the variation is between ingroup (the subtree including all Blechnum) and outgroup (the other subtree).
"Vero" is still quite congruent, so the model used here can't be too much different, either.
What should ring one's alarm bells are, however, the many grade-like / staircase subtrees, which are unusual for a molecular data set. Staircases imply that each subsequent dichotomous speciation event resulted in a single species and a further diversifying lineage: multiple, consistently occurring budding events.
 |
| The same graph, with arrows showing grade evolution. Often found in morpho-data-based trees with ancestral, more ancient, and derived (from them), modern forms, but should ring an alarm bell when common in a molecular tree. Major clades (found in all three trees) are labelled for comparison with the next graph. |
Let's compare this to the Neighbor-net (usually, I would use model-based distances in such a case, but here we can do with uncorrected
p-distances).
 |
| A Neighbor-net inferred from uncorrected p-distances based on Gaspar & Almeida's matrix; the major clades are labelled as in the preceding graph. Note the isolated, long-branch blue dots with asterisks, indicating the position of the first diverged species in the large clades G and I. Genuine signal or missing data artefact? |
The Neighbor-net shows only a limited number of tree-like portions, but does correspond with the main clades above. Only A and B are dissolved, which are the two first diverging clades in the original trees (preceding graph). Some OTUs are placed close to the centre of the graph, or even along a tree-like portion (purple dots), a
behaviour known from actual ancestors: some OTUs apparently have sequences that may be literally
ancestral to others. This explains the grade structure seen in the
original trees. Others (violet dots) create boxes, which may reflect a genuine
ambiguous signal, or just be missing data leading to ambiguous pairwise distances. The latter (missing data artefact) is behind the
misplacement of the four OTUs (red dots): missing data can inflate
pairwise distances severely. And, like parsimony, distance-based methods
are more vulnerable to long-branch(edge)-attraction than probabilistic
methods.
Model-based distances may help clean up this a bit, but the networks needed for these kind of data are Support consensus networks (see e.g.
Schliep et al., MEE, 2017). The split appearance of the Neighbor-net hints at internal signal conflict and, with respect to the high number of variable sites (note the sometimes extremely long terminal edges), saturation issues. Two major questions would be:
- How do the different markers (coding gene vs. inter-genic spacers with different levels of diversity; rps4-trnS is typically more divergent than the trnL-trnF spacer) resolve relationships, which clades / topological alternatives receive unanimous support?
- Does it make a difference to run a fully partitioned (ML) analysis vs. an unpartitioned one vs. one excluding the 3rd codon position in the gene?
For intra-clade evolutionary pathways, it would be worthwhile to give median networks and suchlike a try, as parsimony methods that
can discern ancestor-descendant relationships.
The second molecular dataset
The most recent data are from
Kuo et al. (2017; submission S20277), who inferred a "robust ... phylogeny" (see Part 1, Jamieson et al. 1987, and Beutel et al., above) for a group of ferns, focusing on the taxonomy of a single genus,
Deparia, that now includes five traditionally recognized genera. In the abstract it says: "... seven major clades were identified, and most of them were characterized
by inferring synapomorphies using 14 morphological characters".
The matrix includes the molecular characters used to infer the major clades plus two trees, labelled "bestREP1" and "rep9BEST", both with branch lengths. Branch length values indicate that "bestREP1" could be parsimony-optimized (with averaged or weighted branch lengths), while "rep9BEST" is either a ML or Bayesian tree (technically, it could be a distance-based tree, too, but I don't think such "phenetics" are condoned by
Cladistics).
Re-calculated, the first tree ("bestREP1") is shorter (3024 steps) than the one of Gaspar & Almeida, reflecting the much lower number of parsimony-informative sites (979). Many of the sites differ only between the focal genus and the outgroups, which is well visible in the Neighbor-net. [For those of you unfamiliar with Neighbor-nets, a parsimony analysis of these data takes hours, or days depending on the software and computer, while the distance matrix and the resultant Neighbor-net is inferred in a blink.]
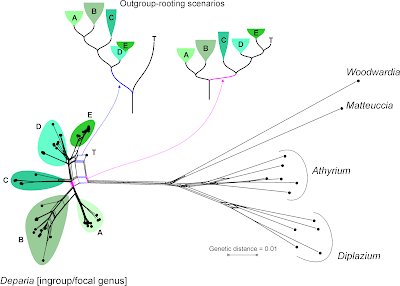 |
| The Neighbor-net based on Kuo et al.'s data. Why do we need to include long-branching, distant outgroups when we just want to bring order in a genus? Because to test monophyly, we need a rooted tree (ambiguous or not, or even biased by branching artefacts). |
Let's remove the distant, long-branching outgroups, which (as we can see in the Neighbor-net) at best provide ambiguous signal for rooting the ingroup — at worst, they trigger ingroup-outgroup branching artefacts. What could a Neighbour-net have contributed regarding taxonomy and the seven major monophyletic intrageneric groups ("clades")? Pretty much everything needed for the paper, I guess (judging from the abstract).
 |
| Same data as above, but outgroups removed. The structure of this Neighbour-net allows to identify seven likely candidates for monophyla ("1"–"7"), with "1" and "2" being obvious sister lineages. Colours refer to the clusters ("A"–"E") annotated above. |
On a side note: by removing the long-branching, distant outgroups, taxon "T" is resolved as a probable member of
the putative monophyletic group "5" (= "E" in the full graph with outgroups, and surely a high-supported subtree in any ingroup-only reconstruction, method-independent). Placing the root between "T" and the rest
of the genus implies that "5" is a paraphyletic group comprising species that haven't evolved and diversified at all (ie. are genetically primitive), in stark contrast to the other main intra-generic lineages. This is not impossible, but quite unlikely. More likely is the second scenario (primary split between "1"–"3" and "4"–"7"). Having "4" as sister to the rest could be an alternative, too.
This is where Hennig's logic could be of help: find and tabulate putative synapomorphies to argue for a set and root that makes the most sense regarding morphological evolution and molecular differentiation.
The take-home message(s)
We have argued before that it is in the ultimate interest of science and scientists to give access to phylogenetic data. No matter where one stands regarding phylogenetic philosophy, we should publish our data, so that people can do analyses of their own. Discussion should be based on results, not philosophies.
When you deal with morphological data, you should never be content with inferring a single tree (parsimony or other).
You have to use networks.
The Neighbor-net was born as late as 2002
(
Bryant & Moulton, 2002, in: Guigó R, and Gusfield D, eds,
Algorithms in Bioinformatics,
Second International Workshop, WABI, p. 375–391; paywalled) and made known to biologists in 2004 (
same authors, same title, in
Mol. Biol. Evol. 21:255–265), so that authors before this time did not have access to its benefits. Similarly, Consensus networks arrived around about the same time (Holland & Moulton 2003, in: Benson G, and Page R, eds,
Algorithms in Bioinformatics: Third International Workshop, WABI, p. 165–176). However, the
Genealogical World of Phylogenetic Networks has been here for six years now (first post
February 2012). So there is now no excuse for publishing a cladogram without having explored the tree-likeness of your matrix' signal!
Neighbor-nets like the ones I showed in this 2-piece post (or can be found in many of our other posts) are a quick and essential tool to explore the basic signal in your matrix:
- How tree-like is it?
- Where are the potential conflicts, obscurities?
- What are the principal evolutionary alternatives (competing topologies)?
- What is well supported (especially regarding taxonomy and the question of monophyly)?
Even if you don't use it in your paper, the network will tell you what you are dealing with when you start inferring trees.
The second essential tool is the much under-used
Support consensus network, not shown in this post but in plenty of our other posts (and many papers
I co-authored; for a comprehensive collection of network-related literature see
Who's who in phylogenetic networks by Philippe Gambette). Support consensus networks estimate and visualize the robustness of the signal for competing topological (tree) alternatives.
Consensus networks should also be obligatory for those molecular data,where even probabilistic methods fail to find a single fully resolved, highly supported tree.
If the editors of
Cladistics are really dedicated to parsimony, they should not still insist only on a parsimony tree (often provided as cladogram), but also parsimony-based networks as well:
- strict Consensus networks to summarize the MPT samples instead of the standard strict Consensus cladograms;
- bootstrap Support consensus networks showing the signal strength and support for alternative trees/competing clades (TNT has many bootstrapping options to play around with); and
- Median networks and such-like for datasets with few mutations, and low levels of expected homoplasy.
This is what the 2016 #parsimonygate uproar (see Part 1) should have been about (12 years after Neighbor-nets, and 11 years after Consensus networks). Not the prioritizing of parsimony, but the naivety or ignorance towards pitfalls of (parsimony or other) trees inferred from data not providing tree-like signal or riddled by internal conflict.
This is a problem not limited to
Cladistics, but found, to my modest experience in professional science (c. 20 years), in many other journals as well
(e.g.
Bot. J. Linn. Soc., Taxon,
Mol. Phyl. Evol.,
J. Biogeogr.,
Syst. Biol., Nature, Science).
Hence, here are my suggestions for future conference buttons, instead of those shown in Part 1.
 |
 |
 |
| No Cladograms! |
Use Neighbour-nets! |
Support Consensus Networks as obligatory! |
Further reading for those who mistrust trees or become network-curious in general































