This is a joint post by Guido Grimm, Johann-Mattis List, and Cormac Anderson.
This is the second of a pair of posts dealing with the names of domesticated animals. In
the first part, we looked at the peculiar differences in the names we use for cats
and dogs, two of humanity’s most beloved domesticated predators. In this,
the second part (and with some help from Cormac Anderson, a fellow
linguist from the
Max Planck Institute for the Science of Human History), we’ll look at two widely cultivated and early-domesticated herbivores: goats and sheep.
Similar origins, but not the same
Both goats and sheep are domesticated
animals that have an explicitly economic use; and, in both cases, genetic and
archaeological evidence points to the Near East as the place of
domestication (Naderi et al. 2007). The main difference between the
two is the natural distribution of goats (providing nourishment and
leather) and sheep (providing the same plus wool). This distribution is also
reflected in the phonetic (dis)similarities of the terms used in our
sample of languages (Figures 1 and 2).
Capra aegagrus, the
species from which the domestic goat derives, is native to the
Fertile Crescent and Iran. Other species of the genus, similar to the
goat in appearance, are restricted to fairly inaccessible areas of
the mountains of western Eurasia (see Figure 3, taken from Driscoll
et al. 2009). On the other hand,
Ovis aries, the sheep and its non-domesticated
sister species, are found in hilly and mountainous areas throughout
the temperate and boreal zone of the Northern Hemisphere. Whenever
humans migrated into mountainous areas, there was the likelihood of
finding a beast that:
Had wool on his
back and hooves on his feet,
Eating grass on a mountainside so
steep
[Bob Dylan: Man
Gave Names to all those animals].
Goats
Goats were actively propagated by
humans into every corner of the world, because they can thrive even in
quite inhospitable areas. Reflecting this, differences in the terms
for "goat" generally follow the main subgroups of the Indo-European
language family (Figure 1), in contrast to "cat", "dog",
and "sheep". From the language data, it seems that
for the most part each major language expansion, as reflected in the
subgroups of Indo-European languages, brought its own term for
"goat", and that it was rarely modified too much or
borrowed from other speech communities.
There is
one exception to this, however. The terms in the Italic and Celtic
languages look as though they are related, coming from the same
Proto-Indo-European root,
*kapr-, although the initial /g/ in
the Celtic languages is not regular. In Irish and Scottish Gaelic,
the words for "sheep" also come from the same root. In other cases,
roots that are attested in one or other language have more
restricted meanings in some other language; for example, the Indo-Iranic words for goat are
cognate with the English
buck, used to designate a male goat
(or sometimes the male of other hooved animals, such as deer).
The German word
Ziege sticks
out from the Germanic form
gait- (but note the
Austro-Bavarian
Goaß, and the alternative term
Geiß,
particularly in southern German dialects). The origin of the German
term is not (yet) known, but it is clear that it was already present
in the Old High German period (8th century CE), although it was not
until Luther's translation of the Bible, in which he used the word,
that the word became the norm and successively replaced the older
forms in other varieties of Germany (Pfeifer 1993: s. v. "Ziege").
 |
| Figure 1: Phonetic comparison of words for "goat" |
Sheep
The terms for sheep, however, are
often phonetically very different even in related languages. The
overall pattern seems to be more similar to that of the words for
dog
– the animal used to herd sheep and protect them from
wolves. An interesting parallel is the phonetic similarity between
the Danish and Swedish forms
får (a word not known in other
Germanic languages) and the Indic languages. This similarity is a
pure coincidence, as the Scandinavian forms go back to a form
fahaz-
(Kroonen 2013: 122), which can be further related to Latin
pecus
"cattle" (ibd.) and is reflected in Italian
[pɛːkora
]
in our sample.
This example clearly shows the limitations of pure
phonetic comparisons when searching for historical signal in
linguistics. Latin
c (pronounced as
[k
])
is usually reflected as an
h in Germanic languages,
reflecting a frequent and regular sound change. The sound
[h
]
itself can be easily lost, and the
[z
]
became a
[r
]
in many Scandinavian words. The fact that both Italian and Danish plus
Swedish have cognate terms for "sheep", however, does not
mean that their common ancestors used the same term. It is much more
likely that speakers in both communities came up with similar ways to
name their most important herded animals. It is possible, for
example, that this term generically meant "livestock", and
that the sheep was the most prototypical representative at a certain
time in both ancestral societies.
Furthermore, we see substantial
phonetic variation in the Romance languages surrounding the
Mediterranean, where both sheep and goats have probably been
cultivated since the dawn of human civilization. Each language uses a
different word for sheep, with only the Western Romance languages
being visibly similar to
ovis, their ancestral word in Latin,
while Italian and French show new terms.
 |
| Figure 2: Phonetic comparison of words for "sheep" |
More interesting aspects
The wild sheep, found in hilly and
mountainous areas across western Eurasia, was probably hunted for its
wool long before mouflons (a subspecies of the wild sheep) were domesticated and kept as livestock.
The word for "sheep" in Indo-European, which we can safely
reconstruct, was
h₂ owis,
possibly pronounced as
[xovis
],
and still reflected in Spanish, Portuguese, Romanian, Russian,
Polish. It survives in many more languages as a specific term with a
different meaning, addressing the milk-bearing / birthing female sheep. These include
English
ewe, Faroean
ær (which comes
in more than a
dozen
combinations; Faroes literally means: “sheep islands”),
French
brebis (important
to known when you want sheep-milk based cheese), German
Aue (extremely rare nowadays, having been
replaced by
Mutterschaf "mother-sheep"). In
other languages it has been lost completely.
What is interesting in this context
is that while the phonetic similarity of the terms for "sheep"
resembles the pattern we observe for "dog", the history of
the words is quite different. While the words for "dog"
just continued in different language lineages, and thus developed
independently in different groups without being replaced by other
terms, the words for "sheep" show much more frequent
replacement patterns. This also contrasts with the terms for "goat",
which are all of much more recent origin in the different subgroups
of Indo-European, and have remained rather similar after they
were first introduced.
The reasons for these different patterns of
animal terms are manifold, and a single explanation may never
capture them all. One general clue with some explanatory power,
however, may be how and by whom the animals were used. Humans, in
particular nomadic societies, rely on goats to colonize or survive in
unfortunate environments, even into historic times. For instance,
goats were introduced to South Africa by European settlers to
effectively eat up the thicket growing in the interior of the Eastern
Cape Province. Once the thicket was gone, the fields were then used
for herding cattle and sheep.
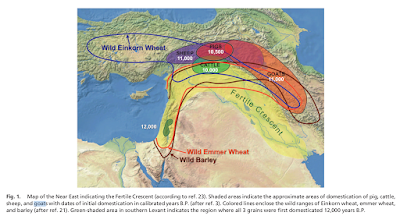 |
| Figure 3: Map from Driscoll et al. (2009) |
There are other interesting aspects
of the plot.
For example, as mentioned before, in Chinese the goat refers to the
"mountain sheep/goat" and the "sheep/goat" is the
"soft sheep". While it is straightforward to assume that
yáng, the term for "sheep/goat", originally only
denoted one of the two organisms, either the sheep or the goat, it is difficult
to say which came first. The term
yáng itself is very old,
as can also be seen from the Chinese character used, which serves as one
of the base radicals of the writing system, depicting an animal with
horns:
羊. The
sheep seems to have arrived in China rather early (Dodson et al.
2014), predating the invention of writing, while the arrival of the
goat was also rather ancient (Wei et al. 2014) (and might also have happened more
than once). Whether sheep
arrived before goats in China, or vice versa, could probably be
tested by haplotyping feral and locally bred populations while
recording the local names and establishing the similarity of words
for goat and sheep.
While the similar names for goat and sheep may be
surprising at first sight (given that the animals do not look all that
similar), the similarity is reflected in quite a few of the world's
languages, as can be seen from the
Database
of Cross-Linguistic Colexifications (List et al. 2014)
where both terms form a cluster.
Source Code and Data
We have uploaded source code and data to
Zenodo, where you can download them and carry out the tests yourself (DOI:
10.5281/zenodo.1066534). Great thanks goes to
Gerhard Jäger (Eberhard-Karls University Tübingen), who provided us with the pairwise language distances computed for his 2015 paper on "Support for linguistic macro-families from weighted sequence alignment" (DOI:
10.1073/pnas.1500331112).
Final remark
As in the case
of cats and dogs, we have reported here merely preliminary impressions, through which we hope to
encourage potential readers to delve into the puzzling world of
naming those animals that were instrumental for the development of human
societies. In case you know more about these topics than we have reported
here, please get in touch with us, we will be glad to learn more.
References
- Dodson, J., E. Dodson, R.
Banati, X. Li, P. Atahan, S. Hu, R. Middleton, X. Zhou, and S. Nan
(2014) Oldest directly dated remains of sheep in China.
Sci Rep 4: 7170.
- Driscoll, C., D. Macdonald,
and S. O’Brien (2009) From wild animals to domestic pets,
an evolutionary view of domestication. Proceedings of
the National Academy of Sciences 106 Suppl 1: 9971-9978.
-
Jäger, G. (2015) Support for linguistic macrofamilies from weighted alignment. Proceedings of the National Academy of Sciences 112.41: 12752–12757.
- Kroonen, G. (2013)
Etymological dictionary of Proto-Germanic. Brill:
Leiden and Boston.
- List, J.-M., T. Mayer, A.
Terhalle, and M. Urban (eds) (2014) CLICS: Database of
Cross-Linguistic Colexifications. Forschungszentrum
Deutscher Sprachatlas: Marburg.
- Naderi, S.,
H. Rezaei, P. Taberlet, S. Zundel, S. Rafat, H. Naghash, et al.
(2007) Large-scale mitochondrial
DNA analysis of the domestic goat reveals six haplogroups with high
diversity. PLoS One 2.10. e1012.
-
Pfeifer, W. (1993)
Etymologisches Wörterbuch des Deutschen.
Akademie: Berlin.
-
Wei, C., J. Lu, L. Xu, G. Liu,
Z. Wang, F. Zhao, L. Zhang, X. Han, L. Du, and C. Liu (2014)
Genetic structure of Chinese indigenous goats and the
special geographical structure in the Southwest China as a
geographic barrier driving the fragmentation of a large population.
PLoS One 9.4: e94435.















