
Homoplasy is the rule, while trait sorting is the exception. Consequently, we have to expect that any morphology-based tree will have more wrong branches than correct ones.
For extant group of organisms, a simple solution to the problem is to analyse morphological traits in the framework of a molecular phylogeny. The genetic data provides us with an independent, best-possible tree. By mapping the morphological traits on this tree, we can evaluate their potency as phylogenetic markers.
But what if our group of organisms is not the product of a simple repeated dichotomous splitting pattern? What if there were anastomoses as well? That is, the morphological traits are not the product of mere (incomplete) lineage or incomplete gene sorting (the latter is called "hemiplasy") but fusion of traits in different lineages. Thus, a tree is not enough to explain the genetic data? What does this imply for the morphological differentiation we observe?
Take the London Plane (Platanus x acerifolia or P. x hispanica), for example, which is a tree that many of us are familiar with. In case you don't know the name: they are the large trees with a patterned bark and deeply lobed leaves and fluffy fruiting bodies found in abundance in parks and alleys throughout the world. It's a cultivation-hybrid (17th—18th century) of the North American plane tree, Platanus occidentalis, and its distant eastern Mediterranean relative, P. orientalis. These are genetically and morphologically distinct species. Their history is summarized in the following doodle (Grimm & Denk 2010).
 |
| Each line represents a semi-sorted nuclear gene region. The split between proto-PNA-E (SW. U.S., NW. Mexico, E. Mediterranean) and proto-ANA (Atlantic-facing Central America, E. U.S.) must have been > 12 myrs ago (last Platanus of Iceland). The minimum air distance between the sister species P. orientalis and P. racemosa is ~11,500 km (via the Arctic). Interestingly, fossils from that time and later (including western Eurasia) have more ANA-clade morphologies: P. orientalis- and P. racemosa-types pop up ~5 Ma. Both ANA and PNA-E clade have distinct morphologies. With respect to the individual gene trees, those exclusively shared by P. palmeri and P. rzedowski with P. occidentalis s.str. and P. mexicana of the ANA clade could be adressed as "hemiplasies". |
If you look at the leaves and fruits of London Planes, you can find everything in between the two endpoints; and the same holds for their genetics. The London Plane is much hardier than Europe's own P. orientalis and more drought-resistant than its hardier North American parent. With climate change going on, the hybrid will eventually meld with the European species entirely. And, thanks to what we call "hybrid vigor", given a few millions of years, it might consume its other parent, too. London Planes have been re-introduced into the Americas; and P. orientalis has become an invasive species in California, where it has started to hybridize with its local sister species P. racemosa. Now imagine a future researcher of Platanus evolution having to deal with a highly complex accumulation of Platanus fossils in the Northern Hemisphere, while being able to study only the left-over complex genetics of a single species that replaced two.
This is where a recently coined new concept comes in: xenoplasy.
Yaxuan Wang, Zhen Cao, Huw A. Ogilvie, Luay Nakhleh (2020). Phylogenomic assessment of the role ofhybridization and introgression in trait evolution. bioRxiv doi: 10.1101/2020.09.16.300343Xenoplasies are traits that originate from hybridization and subsequent introgression. In standard phylogenetics, they would act like any homoplasious character, but their distinction is that they are not independently involved. They are captured via lineage crossing, and reflect a common ancestry.
 |
| Example for a trait incongruent with the species tree, representing a xenoplasy obtained by introgression of I1-A lineage which evolved the trait into I3-B lineage, part of the I2 clade. Pending how far they are affected by incomplete lineage sorting (ILS) and introgression, individual gene may result in any of the three possible genealogies. Modified after Wang et al. (2020), fig. 1. |
As such, their phylogenetic weight (information content) equals that of the anyhow rare classic autapomorphies or synapomorphies (fide Hennig), and this weight is higher than that of the more common homoiologies, shared apomorphies or symplesiomorphies. Note, in the palaeozoological cladistic literature, sorted versions of the latter three are often called synapomorphies – any lineage-specific, derived trait ("synapomorphy") may be lost / modified in some sublineage(s), or rarely pop-up outside the lineage.
Wang et al. provide an analytical framework for identifying a trait as xenoplasy, and assessing the probability for it ("xenoplasy risk factor"). If you're interested in the mechanics, check out the pre-print. The mathematical part of my brain has been dormant for most of the last two decades (when I exchanged chemistry for geology-biology), so I'm more into possible applications to explore this new concept.
Where to look next
The Wang et al. real-world example (Jaltomata) is, however, not very appealing. The problem is that, to look for xenoplasy, we need data that requires us to infer an explicit phylogenetic network (in the strict sense) to start with. In addition, we could use a morphological partition: scored morphological traits; which is usually absent. Last, identifying xenoplasies would make most sense for traits that can be traced in the fossil record, not only to identify potential products of past reticulation but have a better grip on placing critical fossils. Often overlooked by neontologists, fossils are the only physical proof that a lineage was at a certain place at a certain point in time. So, here's two examples: beeches and bears.
Beeches are a small genus of extra-tropical angiosperm trees with a pretty well understood fossil record. Morphologically, their differentiation is very hard to put into a tree, as shown here.
 |
| A morpholgy-based Neigbor-net of fossil (open circles) and extant beech (closed circles) taxa. Coloration gives the (paleo-)geographic distribution (abbreviated as three letters). For more background and information see my Res.I.P. post: The challenging and puzzling ordinary beech – a (hi)story |
Mapping species-discriminating traits on a tree would be of little help here, because the modern species are the product of recurrent phases of mixing and incomplete sorting. I have summarized this in the following doodle, depicting the diversification and propagation of 5S-IGS variants (a non-transcribed, poly-copy, multi-array intergenic nuclear spacer) in a still very small sample.
 |
| A doodle summarizing differentiation patterns in a sample of 686 "representative" 5S-IGS variants obtained using high-throughput sequencing of six beech populations of western Eurasia and Japan (Simone Cardoni et al., to be submitted in the near future; see Piredda et al. 2020 for a similar analytical set-up). |
The people involved in researching this project (drawn by passion rather than resources) don't have the resources to generate the NGS data needed to construct a species network for all of the species of beech, like Wang et al.'s Jaltomata data. But given that there are only 9–10 species, it would be easy prey for a well-funded research group. If you are interested, but don't know how to get the material and are unfamiliar with beeches, feel free to contact the senior author of Piredda et al. 2020, Marco Simeone — new beech-enthusiasts are always welcomed by this group.
Bears are one of the best-studied extant mammal predators, and they also have a decent fossil record. This is probably the reason that Heath et al. (2014) used bears as the case study when introducing their new molecular dating approach: the fossilized-birth-death dating.
 |
| A fossilized birth-death dated tree of bears (modified from Heath et al. 2014, fig. 4). The numbers in brackets give the number of fossil taxa (extinct genera, Ursus spp.) listed on Wikipedia. |
As nice as it looks (and done), their analysis is pretty flawed from an evolutionary point of view. Their dated tree only reflects a single aspect of bear evolution and may involve branch-length artifacts. Heath et al. relied on complete mitochondrial genomes, which they combined with a single nuclear protein-coding gene. Mitochondrial genes reflect only the maternal lineage; they did not date a species tree but a mitochondrial genealogy. Paternal and biparentally inherited gene markers (which includes nuclear genes) tell very different stories about species relationships (this is why we also used the bears as example data for Schliep et al. 2017).
 |
| Strict, branch-length ignorant Consensus network of three trees inferred using species-consensus sequences generated from three sets of data: biparentally inherited nuclear-encoded autosomal introns (ncAI), paternally inherited Y-chromosomes (YCh) and maternally inherited mitochondrial genes (complete set; mtG). This is clearly not the product of a strictly dichotomous evolution. Thick lines: edges found in Heath et al.'s chronogram (= mitochondrial genealogy). |
And while it may be that morphology reflects more the maternal than the paternal side, it has never been tested. Neither how morphology fits with the coalescent species tree. Which would be a network, as shown below.
 |
| Gene flow in bears within the last 5 myrs (estimate; from Kumar et al. 2017). |
How Heath et al. linked the fossils to clades might have been just as wrong as it was right (note that FBD dating is much less biased by mis- or unoptimal placed fossils than traditional node dating). Hemi- and xenoplasy must be considered here. In addition to the highly incongruent paternal and maternal genealogies, we know that even the morphologically most distinct sister species (grizzlies, a special form of Brown Bear, and polar bears) can produce vital offspring ("Grolar") with morphological traits from either side of the family (usually, the Grizzly-side dominates).
Wildlife services usually kill these hybrids as they are considered to speed up the decline of polar bears (they are food competitors). However, with the (possibly inevitable) melting of the polar caps, these hybrids could be instrumental in the survival of a bit of Polar Bear legacy, in the form of genetic diversity not found in brown bears, and xenoplasies. If two highly distinct bear species hybridize today in the wild due to (in this case: human-induced) environmental pressure, their ancestors probably have done so in the past in reaction to shifting habitats and migration patterns.
Given how long bears have intrigued researchers, there are plenty of classic morphological studies involving fossils; and, in the light of the vast amount of molecular data (including ancient DNA!) that have been collected for bears, it should be pretty easy to apply Wang et al.'s new approach to bears. For example, is the Cave Bear a dead-end side lineage, intrograde or hybrid dead-end? Mitochondrial-wise Cave bears are placed as sister to Brown and Polar bears but that's just because of their provenance. Like chloroplast genealogies in plants, mitochondrial genealogies in animals typically show a strong geographic correlation. Especially in bears, the mothers and daughters don't migrate as much as the fathers and sons.
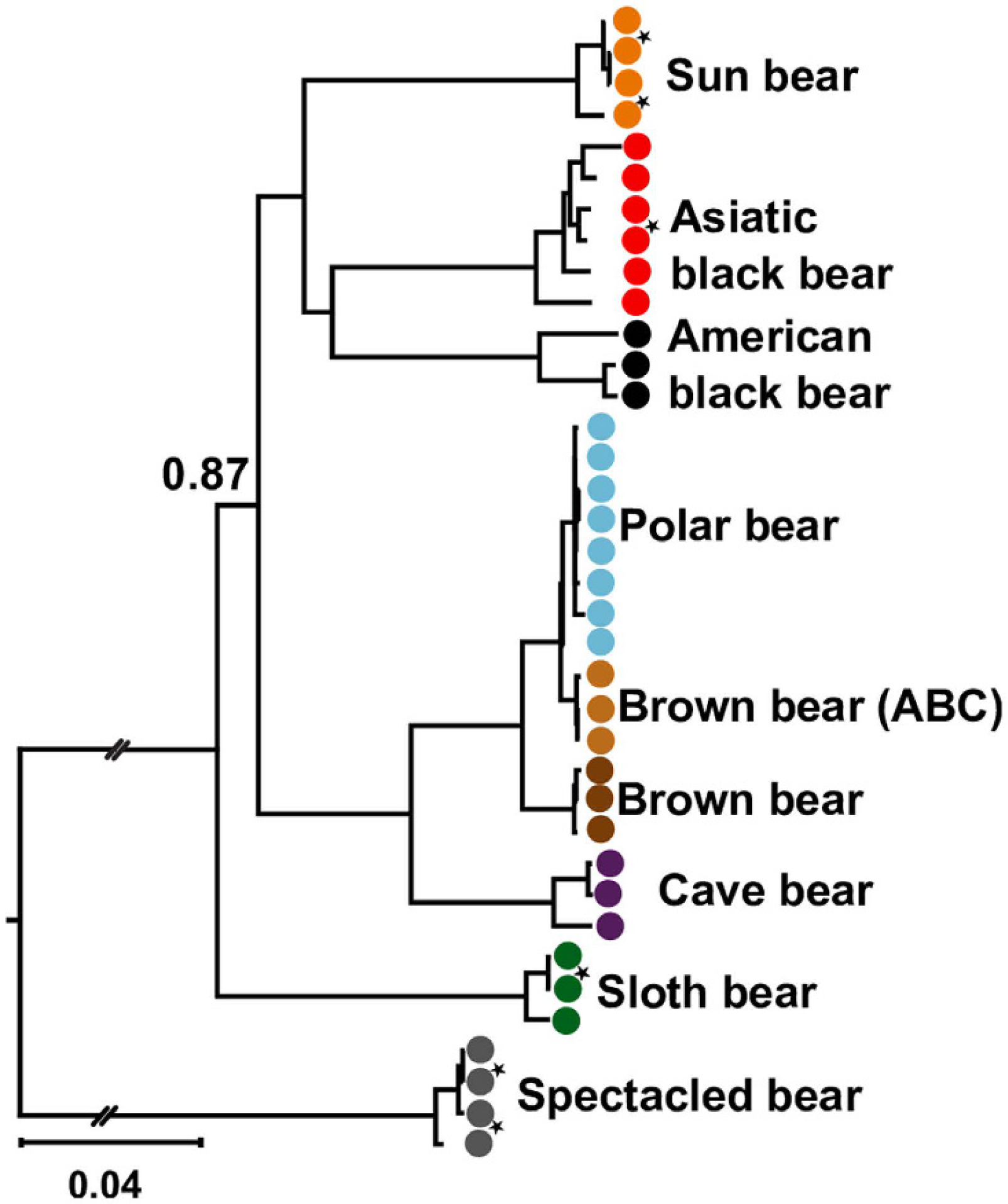 |
| Mitochondrial genealogy of bears including Cave bears (Kumar et al. 2017, fig. 3), the famous European bears of the Ice Ages. ABC bears are insular brown bears living on the subarctic Admirality, Baranof and Chichagof islands of the Alexander archipelago known as natural example for gene flow between Brown and Polar bears (Kumar et al. 2017, fig. 1, provides a map of current distribution of bears). |
Postscriptum
Birds are another animal group that likes to diversify into many species, some of which love to transgress recently established species barriers, forming hybrid swarms. These are actually dinosaurs, a group exclusively studied using cladistic analyses of morphological traits providing non-tree-like signals — mostly homoplasies, a lot of not-really-synapomorphies (good deal are probably homoiologies), and, it wouldn't surprise me, one or another xenoplasy. Or can we assume they were much to advanced to hybridize and intrograde?
Cited literature
- Grimm GW, Denk T. 2010. The reticulate origin of modern plane trees (Platanus, Platanaceae) - a nuclear marker puzzle. Taxon 59:134–147.
- Heath TA, Huelsenbeck JP, Stadler T. 2014. The fossilized birth–death process for coherent calibration of divergence-time estimates. PNAS 111:E2957–E2966.
- Kumar V, Lammers F, Bidon T, Pfenninger M, Kolter L, Nilsson MA, Janke A. 2017. The evolutionary history of bears is characterized by gene flow across species. Scientific Reports 7:46487 [e-pub].
- Piredda R, Grimm GW, Schulze E-D, Denk T, Simeone MC. 2020. High-throughput sequencing of 5S-IGS in oaks: Exploring intragenomic variation and algorithms to recognize target species in pure and mixed samples. Molecular Ecology Resources doi:10.1111/1755-0998.13264.
- Schliep K, Potts AJ, Morrison DA, Grimm GW. 2017. Intertwining phylogenetic trees and networks. Methods in Ecology and Evolution 8:1212–1220.
No comments:
Post a Comment