
Phylogenetic networks are intended to display reticulate evolutionary histories, rather than strictly divergent or transformational histories. This idea applies both to species and higher taxa (where the ancestors might be inferred), and to individuals and populations (where some of the ancestors might be sampled). However, the literature is still replete with studies that use one or more phylogenetic trees for displaying reticulate phylogenies.
A recent example is shown by: Umer Chaudhry, Elizabeth M. Redman, Muhammad Abbas, Raman Muthusamy, Kamran Ashraf, John S. Gilleard (2015) Genetic evidence for hybridisation between Haemonchus contortus and Haemonchus placei in natural field populations and its implications for interspecies transmission of anthelmintic resistance. International Journal for Parasitology 45: 149-159.
These authors sampled nematode parasites from sheep, goats, cattle and buffaloes at abattoirs in Pakistan and southern India. These parasites were morphologically characterized as being predominantly either Haemonchus contortus or Haemonchus placei. The worms were then genotyped in several ways, including: SNPs of rDNA ITS-2, microsatellite markers, sequences of nuclear isotype-1 of β-tubulin, and sequences of mitochondrial NADH dehydrogenase subunit 4. The genotyping revealed several individual worms that were considered to be inter-species F1 hybrids.
The phylogenetic tree from the β-tubulin sequences is shown in the first figure. There were 25 haplotypes identified among the worms. Most of the worms were homozygous, with haplotypes that were identified as either H. contortus or H. placei. However, five worms were discovered to be heterozygous, with one haplotype considered to have come from each of the species.

The hybrid status of the worms is shown in the phylogenetic tree by having the hybrids appear twice, once for each of their haplotypes, with the other worms appearing only once. Thus, the actual reticulate history is not made visually obvious.
A better approach would be to use a phylogenetic network. This is straightforward in this case. From the perspective of the worms (rather than the haplotypes), the phylogenetic tree is a so-called MUL-tree, in which some of the taxon labels appear multiple times (and some appear only once). The labels that appear once represent homozygous worms, which can be seen as being "monoploid" for this locus. The labels that appear twice represent heterozygous worms, which can be seen as being "diploid".
MUL-trees where the labels represent different ploidy levels can easily be turned into a network using the Padre program. The result is shown in the next figure, which is therefore a hybridization network.

The actual history of the worms is now clear. Interestingly, one of the hybridization events seems to be older than the other four.
As an aside, it is also worth pointing out a mis-interpretation of the phylogenetic tree produced from the mitochondrial ND4 sequences. This tree is shown in the next figure — I have added the annotations at the right.
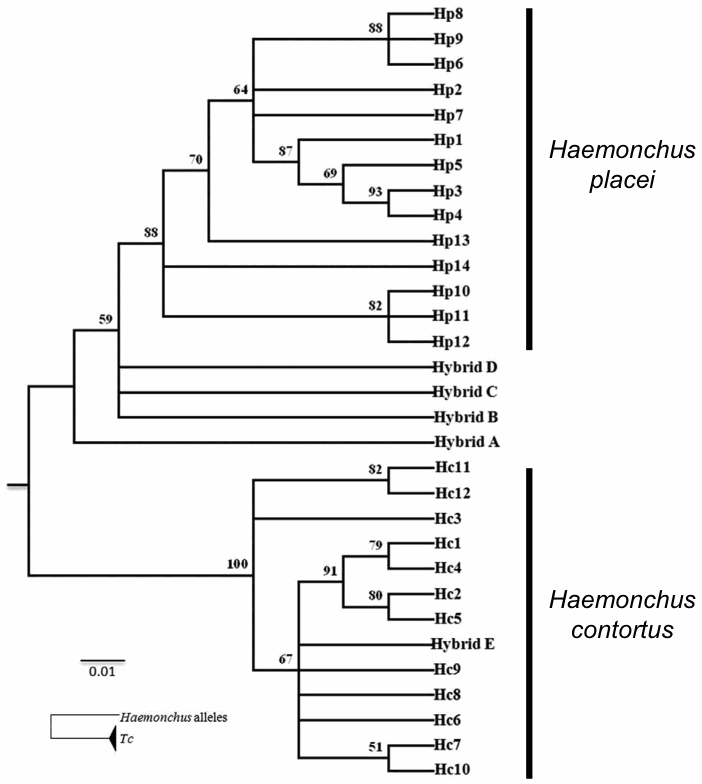
The phylogeny shows 12 haplotypes considered to be H. contortus and 14 haplotypes considered to be H. placei. One of the hybrids clearly has a H. contortus haplotype, indicating that its maternal parent came from this species. However, the other four hybrids cannot be unequivocally identified as having H. placei mothers (as claimed by the authors), as their haplotypes are all sisters to the H. placei haplotypes — all of the H. placei haplotypes share a common ancestor that is not shared with the hybrids. Given the root of the tree, H. placei is a more likely identification than is H. contortus, but the tree does not provide unequivocal evidence.
Informative post. Could any one tell me about the software used for the analysis?
ReplyDeleteThe link to the computer program (Padre) is in the post itself. The program's homepage has a description, as well as a citation for the associated publication. It is straightforward to use — you simply input the multi-labeled tree, and the networks appears immediately. / David
Delete