
I have written before about the interpretation of splits graphs, and provided a simple worked example (How to interpret splits graphs). However, it seems to be worth re-emphasizing the issue here, as I have recently had a paper drawn to my attention that incorrectly infers "groups" of genes from a series of splits graphs.
The essential point to understand is that splits graphs are separation networks. That is, the edges in the graph represent separation between two clusters of nodes in the network; or, they split the graph in two. Formally, each edge (or set of parallel edges) represents a bipartition (or split) of the taxa/genes based on one or more characteristics.
Therefore, the only groups of nodes that are "supported" by a network are those that are represented by splits in the graph, or by some unique combination of splits.
I will illustrate this using the paper already mentioned:
Marz M, Kirsten T, Stadler PF (2008) Evolution of spliceosomal snRNA genes in metazoan animals. Journal of Molecular Evolution 67: 594-607.The authors describe their analyses thus:
We use split decomposition and the neighbor net algorithm (as implemented as part of the SplitsTree4 package) to construct phylogenetic networks rather than phylogenetic trees. The advantage of these method is that they are very conservative and that the reconstructed networks provide an easy-to-grasp representation of the considerable noise in the sequence data.Unfortunately, it is not clear which network algorithm was used for the networks actually presented in the paper. However, this does not affect the interpretation of the graphs (only the number of splits shown).

For Figure 1, the authors claim:
A phylogenetic analysis of the individual snRNA families, nevertheless, does not show widely separated paralogue groups that are stable throughout larger clades. Figure 1, for example, shows that the U5 variants described in Chen et al. (2005) do not form clear paralogue groups beyond the closest relatives of Drosophila melanogaster. On the other hand, there is some evidence for distinguishable paralogues outside the melanogaster subgroup.This interpretation of Figure 1 seems to be quite reasonable.
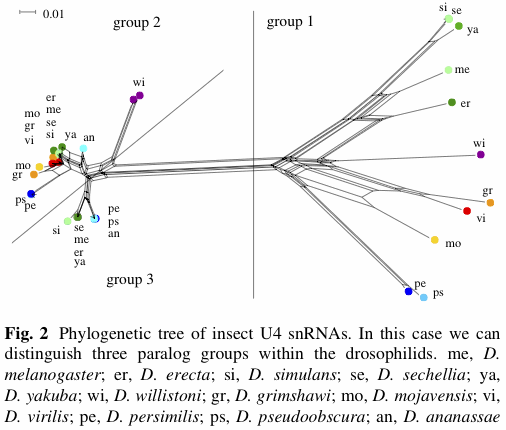
However, for Figure 2 they claim:
The situation is much clearer for the drosophilid U4 snRNAs, where three paralogue groups can be distinguished (see Fig. 2). One group is well separated from the other two and internally rather diverse. The other two groups are very clearly distinguishable for the melanogaster and obscura group (see Drosophila 12 Genomes Consortium 2007). For D. virilis, D. mojavensis, D. grimshawi, and D. willistoni we have two nearly identical copies instead of two different groups of genes.In Figure 2 (which is labelled as a "phylogenetic tree"), only the recognition of "group 1" is very well supported by a split in the network (ie. there is a long set of edges separating the "group 1" genes from the rest of the genes). The distinction between "group 2" and "group 3" does not correspond to any split in the network, although there are a few splits in the network shown that could be used to recognize groups (notably the "wi" genes).

Furthermore, for Figure 3 the authors claim:
In teleost fish, we find clearly recognizable paralogue groups for U2, U4, and U5 snRNAs. Surprisingly, the medaka Oryzias latipes has only a single group of closely related sequences, despite the fact that for U4, the split of the paralogues appear to predate the last common ancestor of zebrafish and fugu (Fig. 3).However, in Figure 3: the left-hand network shows three lines that allegedly define groups, only two of which are supported by splits; the middle network shows three lines that define groups, only one of which is supported by a split; and the right-hand network shows two lines that define groups, neither of which is supported by a split. Once again, there are splits in these networks that do form groupings. For example, in the third network, one of the largest splits supports a grouping of the "bfl" genes, while the other supports a grouping of "bfl" + "pma".
Thus, it seems that the authors' recognition of various paralogue groups is at not well supported by their network analyses. Nevertheless, there are reasonably well-supported splits in the networks shown, which therefore could be used to recognize groups, if desired.
No comments:
Post a Comment