
Constructing phylogenetic networks in situations where introgression or HGT have occurred has been somewhat different in practice to that used for hybridization. Hybridization has usually been tackled by merging incongruent tree topologies, based on the idea that the different topologies represent the phylogenetic history of the different genomes of the hybrid taxon. Introgression and HGT have usually been tackled by adding reticulation edges to a phylogenetic tree, on the basis that the tree represents the phylogenetic history of the main part of the genome.
So, the study of introgression (and HGT) involves (a) constructing a phylogenetic tree from some genomic sample, and (b) detecting the introgressed (or HGT) parts of the genome. This is potentially a problematic procedure, because how do we construct a phylogenetic tree from data that already contain non-tree components? Apparently, the expectation is that a single tree will be supported by the majority of the data, and the remainder will represent the introgressed (or HGT) pathways(s), plus whatever other components have created the observed genomic variability (such as incomplete lineage sorting, gene duplication-loss, and stochastic mutations).
Recently, there have been quite a few studies published that have adopted a specific protocol for this procedure, usually under the rubric of admixture. Most of these have involved the study of ancient human DNA, but there have also been studies of contemporary humans, as well as ancient non-humans, An example of the latter is shown in the next two figures, which represent parts (a) and (b), respectively. They are taken from this study of the relatives of horses: Hákon Jónsson, et alia (2014) Speciation with gene flow in equids despite extensive chromosomal plasticity. Proceedings of the National Academy of Sciences of the USA 111: 18655-18660.

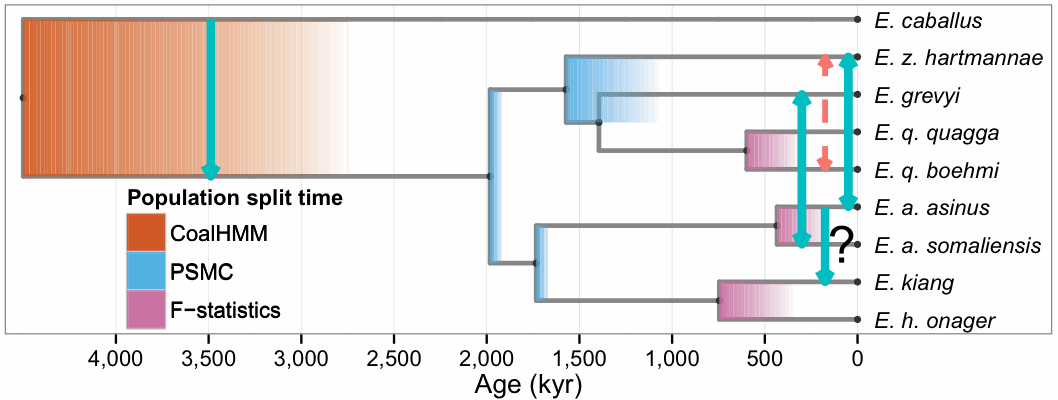
The phylogenetic tree (step a) was constructed using "maximum likelihood inference and 20,374 protein-coding genes ... based on a relaxed molecular clock." So, only stochastic mutations were accounted for when constructing the tree, and not incomplete lineage sorting or gene duplication-loss.
The detection of introgression (step b) used "the D statistics approach, which tests for an excess of shared polymorphisms between one of two closely related lineages (E1 or E2) and a third lineage (E3)". The reticulations representing the detected gene flow were then added to the tree manually.
The D-statistic is also known as the ABBA-BABA test (see: Patterson NJ et alia. 2012. Ancient admixture in human history. Genetics 192: 1065-1093). It operates as follows for sets of four taxa, applied to character data.
Let the species tree be this, where E1–E3 are the three taxa being compared, and O is the outgroup:

There are three possible allele trees for each binary character (ie. single nucleotide polymorphism) in which states are shared pairwise:

In the first tree, E3 shares the ancestral character state with the outgroup, which is expected to be the most common pattern in the absence of gene flow. E1 and E2 share the ancestral state with the outgroup in the second and third trees, respectively.
The admixture test compares the ABBA tree to the BABA tree. The expectation is that if there has been no introgression then the data support for these two trees should be equal. That is, under the null hypothesis that there is no gene flow between the species (and the underlying species tree is correct), the difference in the expected number of occurrences of the ABBA and BABA patterns should be zero. Deviation from this expectation is statistically evaluated using a jackknife procedure.
When there are more than three ingroup taxa, they are tested in groups of three (plus the outgroup). No correction for multiple hypothesis testing seems ever to be applied. Recently, the test has been extended to five taxa (Pease JB, Hahn MW. 2015. Detection and polarization of introgression in a five-taxon phylogeny. Systematic Biology 64: 651-662).
Note that this test assumes that:
- the "excess of shared polymorphisms" arises solely from gene flow, with or without incomplete lineage sorting, rather than from any other tree-like processes such as gene duplication-loss or ancestral population structure
- there are no other sources of co-ordinated polymorphisms, such as character-state reversals due to adaptation / selection
- any gene flow that does exist is due to introgression, rather than to hybridization or HGT.
No comments:
Post a Comment