
As a means of motivating his interest in speciation, in The Origin of Species Charles Darwin highlighted the diversity of morphological forms among the finches of the Galápagos Islands, in the south-eastern Pacific Ocean, which he visited while circumnavigating the world in The Beagle. He considered this to be a prime example of biodiversity related to adaptation and natural selection, what we would now call an adaptive radiation.
Recently, the following paper, which provides a genomic-scale study of these birds, has attracted considerable attention:
Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A, Promerová M, Rubin CJ, Wang C, Zamani N, Grant BR, Grant PR, Webster MT, Andersson L (205) Evolution of Darwin's finches and their beaks revealed by genome sequencing. Nature 58: 371-375.The authors note:
Darwin's finches are a classic example of a young adaptive radiation. They have diversified in beak sizes and shapes, feeding habits and diets in adapting to different food resources. The radiation is entirely intact, unlike most other radiations, none of the species having become extinct as a result of human activities.
Here we report results from whole genome re-sequencing of 120 individuals representing all Darwin's finch species and two closely related tanagers. For some species we collected samples from multiple islands. We comprehensively analyse patterns of intra- and inter-specific genome diversity and phylogenetic relationships among species. We find widespread evidence of inter-specific gene flow that may have enhanced evolutionary diversification throughout phylogeny, and report the discovery of a locus with a major effect on beak shape.Sadly, the authors try to study the intra- and inter-specific variation principally using phylogenetic trees. They do this in spite of noting that:
Extensive sharing of genetic variation among populations was evident, particularly among ground and tree finches, with almost no fixed differences between species in each group.Clearly, this situation requires a phylogenetic network for adequate study, as a network can always display at least as much phylogenetic information as a tree, and usually considerably more. The authors do recognize this:
A network constructed from autosomal genome sequences indicates conflicting signals in the internal branches of ground and tree finches that may reflect incomplete lineage sorting and/or gene flow ... We used PLINK to calculate genetic distance (on the basis of proportion of alleles identical by state) for all pairs of individuals separately for autosomes and the Z chromosome. We used the neighbour-net method of SplitsTree4 to compute the phylogenetic network from genetic distances.However, this network is tucked away as Fig. 3 in the appendices. It is shown here in the first figure. The authors attribute the gene flow to introgression, but occasionally refer to hybridization and convergent evolution. Indeed, they suggest both relatively recent hybridization as well as the possibility of more ancient hybridization between warbler finches and other finches.

Clearly, this network is not particularly tree-like in places, especially with respect to the delimitation of species based on their morphology, as reflected in their current taxonomy. Nevertheless, the authors prefer to present as their main result as a:
maximum-likelihood phylogenetic tree based on autosomal genome sequences ... We used FastTree to infer approximately maximum-likelihood phylogenies with standard parameters for nucleotide alignments of variable positions in the data set. FastTree computes local support values with the Shimodaira–Hasegawa test.This tree is shown in the second figure.
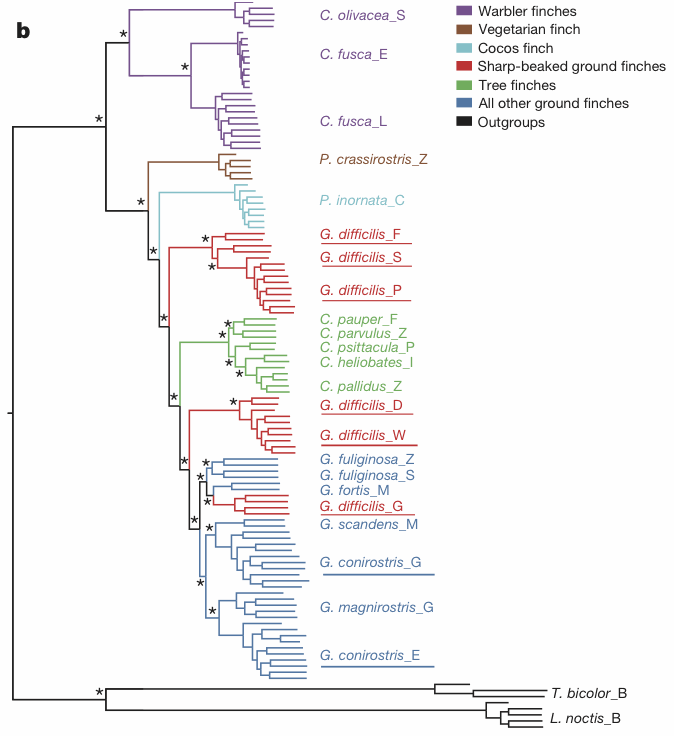
This apparently well-supported tree is not a particularly accurate representation of the pattern shown by the network. Indeed, it makes clear just why it is inadequate to use a tree to study the interplay of intra- and inter-specific variation. Gene flow requires a network for accurate representation, not a tree.
The authors do acknowledge this situation. While they try to date the nodes on their tree, they do note that:
Although these estimates are based on whole-genome data, they should be considered minimum times, as they do not take into account gene flow.Actually, in the face of gene flow the concept that a node has a specific date is illogical, because the nodes do not represent discrete events (see Representing macro- and micro-evolution in a network). Given the authors' final conclusion, it seems quite inappropriate to rely on trees rather than networks:
Evidence of introgressive hybridization, which has been documented as a contemporary process, is found throughout the radiation. Hybridization has given rise to species of mixed ancestry, in the past and the present. It has influenced the evolution of a key phenotypic trait: beak shape ... The degree of continuity between historical and contemporary evolution is unexpected because introgressive hybridization plays no part in traditional accounts of adaptive radiations of animals.
No comments:
Post a Comment