
As phylogeneticists, we commonly have to deal with data that we don't initially understand. In this post, I'll use a recently published 8-gene dataset on lizards to show how much can be learned prior to any deeper analysis, just from producing a few Neighbour-nets.
The data
Solovyeva et al. (Cenozoic aridization in Central Eurasia shaped diversification of toad-headed agamas, PeerJ, 2018) sampled species of toad-headed agamas (lizards) across their natural range (north-western China to the western side of the Caspian Sea), to study their genetic differentiation in time and space. To do so they used two datasets. The mitochondrial data covers four gene regions: coxI, cytB, nad2, and nad4, and are complemented by four nuclear gene regions: AKAP9, NKTR, BDNF, RAG1.
This caught my eye, because the authors' preferred trees have a bunch of low branch-support values, so that this would be a good opportunity to advocate some Consensus networks. They also report only values above a certain threshold, as apparently recommended by several reviewers. My reviewers not rarely recommended the same, but I always ignored this — I believe we should give the value, because it makes a difference if its just below the threshold (e.g. bootstrap support, BS, of 49), or non-existent (BS < 5). The authors also note that their mitochondrial and nuclear genealogies are not fully congruent. In short, the signal from their matrix is probably not trivial, but could be interesting.
In contrast to many other journals, PeerJ has a strict open-data policy. Solovyeva et al. provide each gene as FASTA-formatted alignment as Supporting Information. So let's have some quick-and-dirty Neighbour-nets.
Using Neighbour-nets to decide on an analysis strategy
A comprehensive outgroup sampling can avoid outgroup-rooting artefacts, but adding very distant outgroups comes at a price. We need to invest much more computational effort, because the inference programmes not only try to optimize our focus group, but the entire taxon set. Another principal question is: what can an outgroup taxon provide as information for rooting an ingroup, while being completely different? Furthermore, when we do an ML (or Bayesian) analysis, e.g. with RAxML, we leave it to the program to optimize a substitution model (even when we predefine a model, its parameters will usually be optimized by the inference software on the fly). By adding distant outgroups, we optimize a model for them plus our focus group — by not using any outgroup, we optimize a model suiting just the situation in our focus group.
Fig. 1 shows the neighbour-net (uncorrected, codon-naive p-distances) for the first of the mitochondrial genes, coxI (the others are similar), which and tells us a lot about the data to be used for the tree inferences.
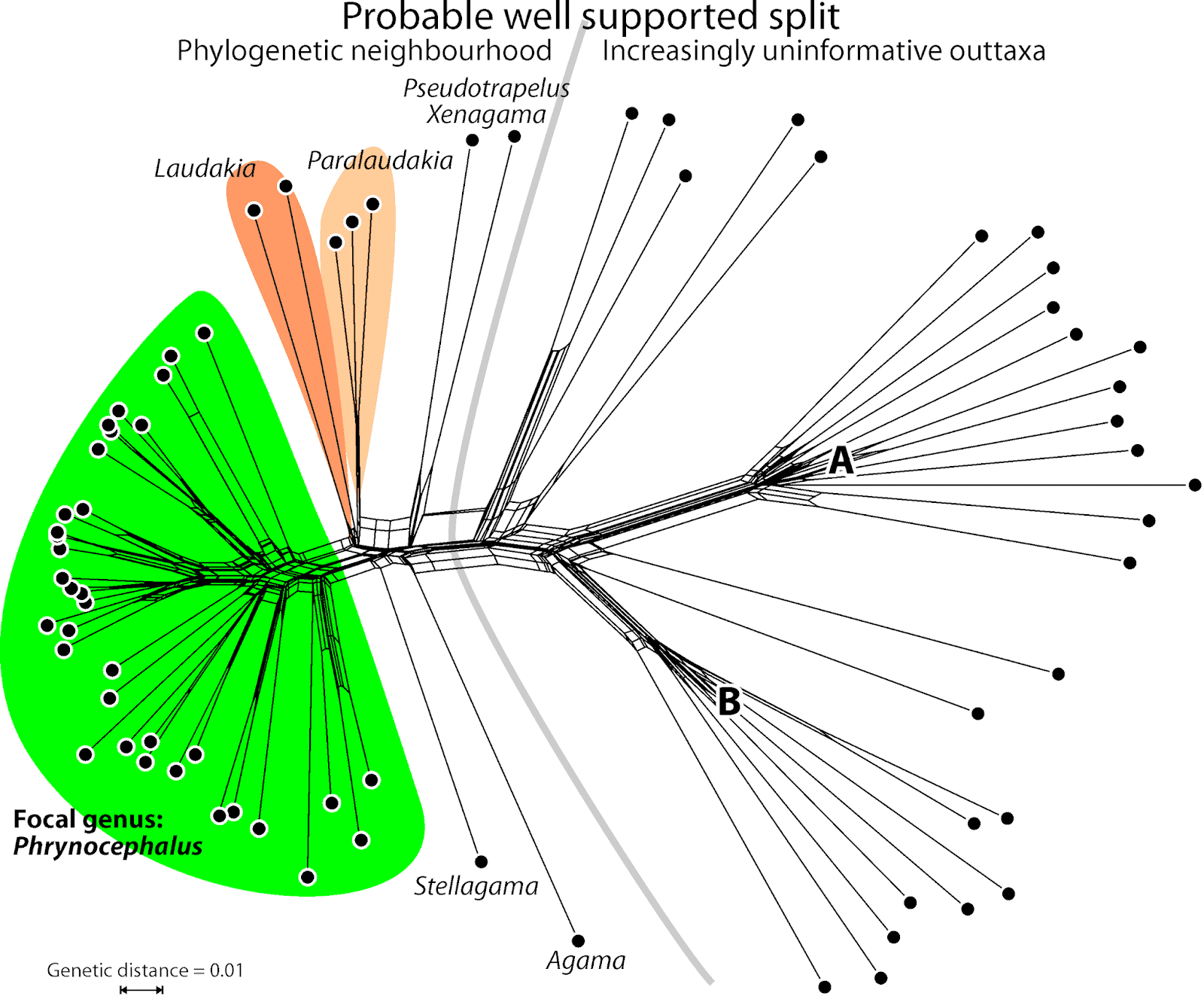 |
| Fig. 1 Neighbour-net based on mitochondrial (coxI) uncorrected p-distances. The diffuse, non-treelike signal expressed in the A and B fans will be a hard nut for the tree inference, and will have little influence on questions dealing with the focal genus. |
We also can see that the 3rd codon position is probably saturated to some degree, and that we will be dealing with a high level of stochasticity (randomly distributed mutation patterns) here — all terminal edges are long to very long. Since the same thing holds for the other three mitochondrial regions, it would not be a bad idea to do an additional inference including only the 1st and 2nd codon positions, in case all taxa should be included.
Using Neighbour-nets to understand the basic signal properties of your data
Fig. 2 shows the Neighbour-net (again, uncorrected p-distances) for one of the nuclear genes, AKAP9. The outgroup sample is somewhat different, but we can immediately see that this gene has more potency to infer unambiguous phylogenetic relationships among the sampled taxa — the graph has distinctly tree-like portions. We also see that saturation of 3rd codon position is much less of an issue here, compared to the cox1 gene (Fig. 1) — the terminal edges are comparatively short, with respect to the central edge bundles. [Nonetheless, it is never wrong to analyze coding gene data partitioned: 1st and 2nd codon positions vs. 3rd codon position.]
 |
| Fig. 2 Neighbour-net based on the nuclear (AKAP9) genetic distances. Note the much more treelike structure of the graph, the generally shorter terminal edges, and last-but-not-least the notable difference between ingroup (focal genus) and outgroup taxa. |
Similarly structured graphs are found for the other three nuclear genes.
Producing some quick Neighbour-nets doesn't hurt
Sometimes reviewers will pick on them — "distance-based phenetic method" is something I used to get a lot. In this case, you can still produce them just to get some basic impressions on your data set. This will help you to understand the results of your tree inferences, including why some of your branches have ambiguous support.
It comes as little surprise that the taxa one can identify, in these networks, as likely sister genera of the focal genus, come up as sister taxa in the explicit phylogenetic analyses done by Soloveya et al. — e.g., their fig. 2 showing the combined mitochondrial tree, and their fig. 3, showing the combined nuclear tree.
Soloveya et al. (2018) performed some incongruence tests (AU-topology test) using single-gene inferences (going further than many other studies), but did not dig deeper. One of the authors answered my question about potential signal issues that may cause topological incongruence between ML and Bayesian trees, as well as ambiguous support, but he considers this to be a solely a problem with methods — different algorithms prefer different phylogenies. Having looked at the basic differentiation pattern in the gene regions using Neighbour-nets, it may be more than just an issue with methods — ML and Bayesian analysis should always support the same splits when using the same or similar substitution models.
Like many other studies, the authors also use the data for Bayesian dating and dating-dependent biogeographic analysis. Lacking any ingroup fossils, the authors could only constrain nodes within the outgroup subtree, which are nodes far from those that they discuss and estimate. I have my doubts that we can put much faith in the uncorrelated clock process to handle such extreme differences between focus group (ingroup) and (constrained) outgroup-taxon lineages as seen in Fig. 2. Estimates for rate shifts between outgroup and ingroup usually render ingroup age estimates to be too young, compared to age estimates obtained with ingroup fossils. This is something that can be directly deduced from a graph like the one in Fig. 2.
Data and networks can be found at figshare
The original paper provides a comprehensive supplement with a lot of interesting information, but the FASTA-files, each comprising a single gene region and a few editing issues, are not yet ready to use. Hence, I transformed them into NEXUS-files, and generated a combined data matrix. The files and the Neighbour-nets for each gene region (and a full single-gene maximum likelihood analysis) can be found on figshare.
No comments:
Post a Comment