
Application of networks should long have been standard in molecular phylogenetics, to get the most out of the available data. However, you will rarely find one in a systematic botanical paper, at low- or high-hierarchical levels. Instead, the focus of systematic botanical research is to just leave branches with ambiguous support aside until somebody puts together the resources to generate phylogenomic data allowing to infer a fully resolved tree.
It therefore is an interesting exercise to take some systematic research and to look at what networks reveal. To this end, using Stevens' (2001 onwards) brilliant webpage, the Angiosperm Phylogeny Website, I will pick some of the low-supported branches, and show what networks could have revealed (in some cases, long ago)
Stevens essentially collects all of the literature on the various taxonomic groups of angiosperms, making it probably by far the best resource to start with when looking into an angiosperm group. He also provides synoptical trees (permanently updated when new evidence comes up) for the angiosperms at a whole, and their sublineages down to the order level, and annotates the level of support for the branches using generalized categories. My first example (personal interest due to my former research) will be the Fagales.
Fagales
Here's Stevens' overview tree (Fig. 1) for this economically very important order.
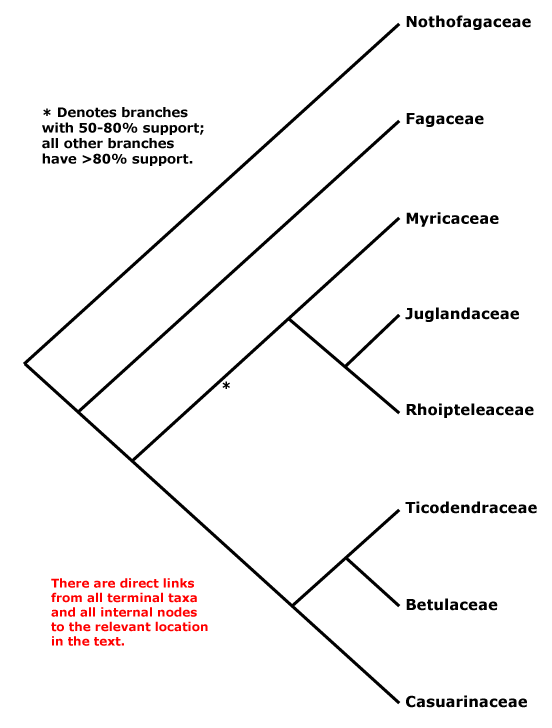 |
| Fig. 1 A phylogenetic synopsis for the order Fagales (Stevens 2001 onwards). Except for the asterisk branch, this topology is consensual among any study including representatives of the families of the Fagales. |
It's a rather small order with just 7–8 families, interfamily relationships "... are fairly well resolved, although the position of Myricaceae remains uncertain", as it has been for more than a decade. The topology in Fig. 1 is the one found by Li et al. (2004), who wrote in their abstract:
Nucleotide sequences of six regions from three plant genomes — trnL-F, matK, rbcL, atpB (plastid), matR (mtDNA), and 18S rDNA (nuclear) — were used to analyze inter- and infrafamilial relationships of Fagales. All 31 extant genera representing eight families of the order were sampled. Congruence among data sets was assessed using the partition homogeneity test, and five different combined data sets were analyzed using maximum parsimony and the Bayesian approach. At the familial level, the same phylogenetic relationships were inferred from five different analyses of these data. Nothofagus, followed by Fagaceae, are subsequent sisters to the rest of the order. Fagaceae are then sister to the core ‘‘higher’’ hamamelids, which consist of two main subclades, one being Myricaceae (Rhoipteleaceae (Juglandaceae)) and the other Casuarinaceae (Ticodendraceae (Betulaceae)). The combined data sets provide the best-supported estimate of evolutionary relationships within Fagales. Our results suggest that the combination of different sequences from several species within the same genus representing a terminal taxon has little influence on phylogenetic accuracy. Inclusion of taxa with some missing data in combined data sets also does not have a major impact on the topology.All solved, it seems. The interesting thing is that the only branch with moderate support relates to those families that have the oldest record. Myricaceae and Juglandaceae pollen types can be found deep into the Late Cretaceous, often classified under the form taxon Normapolles, which also includes pollen morphs of uncertain relationship to modern-day Fagales. Myricaceae and Juglandaceae are short-rooted, in contrast to the two first-diverging families, the enigmatic southern hemispheric Nothofagaceae, the false beech, and the (mostly) northern hemispheric Fagaceae, including the trees every European and American knows: the beech trees and oaks. Without these trees, Brittania would have never ruled the waves — especially widespread oaks provide excellent ship wood.
The basic situation: treelike and non-treelike parts
Li et al. (2004) did not show a phylogram, which is still a standard in systematic botany. The "asterisk" branch may just relate to little discriminative signal. Regarding the root, we always should be cautious regarding ingroup-outgroup long-branch attraction. Molecular data has an inherent dilemma. A group diverging first, and earlier than all others (here: Nothofagaceae), should be genetically most distinct. But a later-diverging but fast-evolving group may be more distinct, and hence attracted to the outgroup, which (naturally) may be very distinct from the ingroup.
We don't need to make a full tree-analysis to become aware of the primary signal issues in Li et al.'s data set, a simple neighbour-net will do (Fig. 2).
 |
| Fig. 2 Neighbour-net based on simple (Hamming) p-distances inferred from Li et al.'s matrix. Alternative roots refer to the 18S rDNA-inferred root and earliest fossil evidence for discrete Fagales lineages. |
From the neighbour-net we can see:
- Rhoiptelea, the only member (1-2 species) of the Rhoipteleaceae is much closer to the Juglandaceae than the beeches (Fagus) are to the remainder of its family ("quercoids" within Fagaceae). Interestingly, is has been suggested to include the Rhoipteleaceae as a subfamily within the Juglandaceae, but no-one has come forward with the idea of splitting the Fagaceae.
- We also see that the ambiguous support for the Juglandaceae (s.l.) + Myricaceae clade is indeed due to a lack of straightforwardly discriminating signal.
- The outgroup-root may be problematic. The Nothofagaceae are most distinct within the order, with little affinity to any other main group. The neighbour-net is a distance-based analysis, and as such vulnerable to long-branching artefacts. Conspicuously, we have an edge bundle pulling one outgroup taxon closer to the equally distinct Fagaceae. But it's impossible to judge whether the outgroup-inferred root is an artefact or not — any outgroup sample (no matter how comprehensive) will enforce the split between the unique Nothofagaceae and the remaining Fagales, and the second-most distinct Fagaceae and the rest.
The signal issues apparent from the neighbour-net find a reflection in the maximum likelihood bootstrap consensus network (Fig. 3).
 |
| Fig. 3 ML-bootstrap consensus network, based on a partitioned analysis (no cut-off); same data than used for Fig. 2. Note that the moderate support for the Myricaceae-Juglandaceae sister relationship (BS = 62, blue) has only one alternative realized in all other BS (pseudo-)replicates (BS = 38, red; 62+38 = 100). |
Now, we know that although there is little discriminating signal, the data is decisive about what to do with the Myricaceae — their position is not "uncertain", but instead there are two alternatives: two-thirds of the segregating sites find that they are sister to the Juglandaceae (s.l.), and the other third place them as sister to the BTC clade including Betulaceae, Ticodendraceae and Casuarinaceae.
From an evolutionary point of view, such a situation is easily explained. The splits between the first ancestors of either clade may have been temporally very close, and affected by incomplete lineage sorting, leading to competing signals. Different evolutionary rates in the BTC and Juglandaceae stem lineages compared to that of the Myricaceae would have made BTC and Juglandaceae distinct from each other, but not from the Myricaceae. Another thing may be that the first Myricaceae was geographically closer to the first Juglandaceae than to the first BTC, so that their plastids were more similar (BS = 62)., even though the evolutionary sequence was: Juglandaceae diverge first, then BTC and Myricaceae splits up (BS = 38, mainly supported by the biparentally inherited 18S rDNA).
Let's check these hypotheses.
Three genomes with four different signals
Li et al., and all studies done afterwards, were sure that there are no issues with incongruence. However, they overlooked the imbalance in gene sampling, and the insufficiency of classic tests to uncover actual incongruence. Furthermore, there is no guarantee for compatible data even when the maternal and paternal genealogies (the true trees) are congruent. Different gene regions may reflect certain aspects of the true tree very well, and mess-up others. This is clearly the case for Li et al.'s data set, as evidenced in their Betulaceae subtree. All of the branches have unambiguous support (Fig. 3), but they are wrong when compared to densely sampled data sets using gene regions with more differentiation potential, close to the leaves of the Fagales tree ("actual Betulaceae subtree" indicated in Fig. 3; cf. Grimm & Renner 2013).
The authors used three coding and one non-coding plastid region, adding one mitochondrial gene (matR) and one nuclear-encoded ribosomal gene, the 18S rDNA, a gene region that had long be known to be sequentially very conservative (thus, easy to sequence). Plastid and mitochondrial signatures are maternally inherited in most plants and all flowering plants (angiosperms) as far as studied; the 18S rDNA is part of a tandemly repeated coding unit, the 35S rDNA cistron, inherited from the paternal and maternal side.
Let's assume that
- all genes contribute equally to the amount of segregating sides, but
- the plastid and mitochondrial regions prefer one topological alternative (A), and the 18S rDNA, being biparentally inherited, prefers another (B).
Any support <<100 (or PP <1.0) may be an indication of incompatible signals and, potentially, conflict. Thus, the only valid test for congruence is to infer single-gene (single-partition) trees, or at least single-genome trees, and then assess whether there are conflicting branches with high support.
Fig. 4 shows the single-gene trees that can be inferred from Li et al.'s matrix, revealing some significant (well-supported, BS > 80) topological incongruence.
 |
| Fig. 4 Single-gene trees for Li et al.'s data set. A. 18S rDNA. B. atpB (plastid gene). C. matK (plastid gene), powerful marker providing phylogenetic backbones in essetially all angiosperm studies above the genus level; can outcompete any conflicing signal from other regions. D. matR; the only mitochondrial gene known for a large range of angiosperms, typically with very little discriminative power, reflected here by overall poor support. E. rbcL (plastid gene), the classic angiosperm marker, provides very stable, relatively deep backbones signals. F. trnL intron and trnL-trnF intergenic spacer, best-sampled (to this day) non-coding plastid gene region; alignment can be tricky beyond family and order level, but typically conserved within families and genera (reason for non-100% barcoding success; closely related genera will usually only differ by few, typically convergent and not rarely stochastically distributed point mutations and indel patterns). |
With respect to the signal issues seen in the networks based on the combined data, each of the single-gene trees, and their phylogenetic prospects and pitfalls, could be discussed. However, I will only highlight some striking aspects here.
- Including matR data to cover "all three genomes" is scientific sham. The region does not provide any useful signal (note the low support in Fig. 4D). For the Fagales, it fails miserably to even find unambiguous, long-known groups. And this probably holds for most other datasets that include this region (see this post on networks helping to identify biased roots). If it has any use at all, it's for very deep splits (the Fagales crown-group radiation goes back at least 80 Ma) or groups with extremely inflated mutation rates. But bewareof the difference between 1st/2nd and 3rd codon position, as the latter shows a lot of stochasticity. With respect to the diffuse signal from this region, it may even be hurtful to include (in particular, when using non-probabilistic inference methods).
- The split support regarding the Myricaceae is indeed due to incongruent nuclear (biparentally) and plastid signals. But it's contrary to the theory that the plastids out-compete the single nuclear gene — the 18S rDNA out-competes partly or fully incongruent signals from the plastid genes! By adding a plastid gene, we only reduce the near-unambiguous (BS = 97, a lot for a single gene) support for a Myricaceae-Juglandaceae clade. It is, however, not enough to bias the situation entirely, because the conserved plastid genes (atpB, rbcL; Fig. 4B, E) provide somewhat diffuse signals. For instance, the atpB-BS = 38 for the Myricaceae as sister to Juglandaceae and BTC clade competes with the 18S-preferred alternative, and the one preferred by the more variable matK plus the non-coding trnL/LF. The rbcL signal does not help too much the matK–trnL/LF case, because it messes up the ingroup by placing the Fagaceae deep within the core clade.
- The nuclear gene prefers a different root. We have a BS = 89 (all other alternatives BS < 5) for a clade comprising all cupuliferous, mostly extratropical Fagales: the southern hemispheric Nothofagaceae and the (mostly) northern hemispheric Fagaceae. Note the comparatively low root-tip distance for Nothofagus in case of the 18S rDNA compared to other Fagales (Fig. 4). This is a signal that is completely wiped out in the combined data (Fig. 3), as all of the other regions provide a near-unambiguous (BS > 90) support for a Nothofagus + outgroup vs. the remaining Fagales split.
Returning to our evolutionary hypothesis, Li et al.'s data (properly analysed) indicates that the slow-evolving (or slower) Myricaceae originated geographically close to the common ancestor of the BTC clade, but at the same time were evolutionarily closer to the Juglandaceae. It is important to keep in mind that back then, in the Late Cretaceous (or earlier), when all three families evolved, any then-existing systematicists would probably have recognised all three ancestors and their precursors as species of the same genus, or at least genera of the same family. This follows the example of modern-day Fagaceae and Nothofagaceae, where the plastids are geographically strongly constrained and largely decoupled from morphology (taxonomy) and nuclear genealogies.
Alternative topologies can be evolutionary clues
The data of Li et al. (2004) may appear to be quite old, but effectively any inference will find the same patterns for the deep relationships in the Fagales.
But there is no need to stop with "asterisk" branches. Networks, even those inferred using tree frameworks (our bootstrap support networks), can illuminate the reasons for ambiguous support. We can put up evolutionary scenarios to explain ambiguous support (not necessarily involving reticulation) or at least we can discuss ambiguous support in an evolutionary context. What are the topological alternatives (here: Myricaceae sister to Juglandaceae or sister to BTC clade)? Which data support which alternative? Are there evolutionary processes such as ancient hybridisation, incomplete lineage sorting, or simply fast radiation, generating such signals? How does this fit with the fossil record (palaeo-distribution in space and time)?
The "asterisk" branches in the angiosperm Tree of Life may be just as relevant for understanding the evolution of a group as the clear ones. Indeed, they may be even more interesting to look at. For sure, they should not be regarded in general as just indecisiveness of available data, i.e. topological uncertainty.
No comments:
Post a Comment