In the last week of May the idyllic mountain retreat Hameau de l'Etoile, in the Montpellier region of France, was the location for the conference Mathematical and Computational Evolutionary Biology 2013. (Well, perhaps 'mountain' is not the correct word, but for somebody based in the Netherlands the difference between a hill and a mountain is a largely academic concept...)
The main theme of the conference was the interface between phylogenetics and health. Phylogenetic networks, in all their various guises, were quite prominent at the conference. During the poster sessions themes such as maximum parsimony on networks, maximum acyclic agreement forests (used for parsimoniously merging two gene trees into a species network) and unrooted, "data display" phylogenetic networks were evident. There were also quite a few posters on parsimonious gene tree species tree reconciliation, which can, in principle, be used to build rooted phylogenetic networks.
Networks were also visible in a number of the long talks. On the first day, I closed the afternoon with an attempt to summarise in (very) broad lines the methodologies available for constructing rooted phylogenetic networks, and the extent to which these techniques have been biologically validated. This, if you remember, is a theme that has surfaced on this blog at least once or twice in the past. In the talk I argued that there is actually more work out there on validation than many people realise, particularly on horizontal gene transfer and to a lesser extent on hybridization. However, differences in the terminology and models used by different research groups means that a lot of this work is overlooked, even when the conceptual similarities are strong (e.g. DLT-reconciliation, Ancestral Recombination Graphs and networks).
On the second day, Tuesday, Bastien Boussau gave a detailed, but very clear, talk about various different flavours of reconciliation. Alongside different combinations of deletion, loss, transfer and incomplete lineage sorting he also looked at the multiple points in the traditional phylogenetic pipeline where uncertainty exists (alignment, species tree construction, gene tree construction, reconciliation) and how these can be co-estimated.
Wednesday saw Gil McVean describe his work on analysing the data harvested by the 1000-genomes project. Here, networks popped up in the form of Ancestral Recombination Graphs (ARGs). ARGs can perhaps best be summarised as "phylogenetic networks for population genetics". Rather than modelling horizontal gene transfer or hybridization, reticulations represent recombination, which requires the character data to have an explicit linear ordering (e.g. SNPs), Gil, wisely, did not try and construct an explicit ARG for the data from the 1000 genome project (think: SNP matrix with 1000 rows and several million columns/SNPs...) although he did describe a number of statistics summarising key properties of it, e.g. the average distance that has to be travelled back in time before two UK citizens reach a common ancestor.
The final talk that explicitly addressed network themes was by Darren Martin. He motivated why one should try and detect recombination, and went into detail about techniques for detecting (and interpreting) recombination in viruses. He explained how his software RDP4 for detecting recombination breakpoints works. As with many other speakers, he devoted part of his talk to HIV.
The full program, and slides for most of the long talks, are available at the following URL. Needless to say, there were also plenty of very interesting talks that had no direct connection with networks.
http://www.lirmm.fr/mceb2013/program.php
There are also detailed abstracts available for all the short talks and posters:
http://www.lirmm.fr/mceb2013/MCEB2013_program.pdf

Biology, anthropology, computational science, and networks in phylogenetic analysis
Wednesday, June 26, 2013
Monday, June 24, 2013
The first Darwinian evolutionary tree
Tassy (2011) has pointed out that a Darwinian evolutionary tree has certain key characteristics that (in combination) distinguish it from other models of evolution, such as those devised by Darwin's predecessors:
- it includes ancestral and descendant forms
- ancestral taxa are species not higher taxa
- extant taxa are only at the leaves not the internal nodes or edges
- there is gradual a transition between forms
- there is splitting of lineages.
Almost all of the early trees and networks do not match at least one of these criteria. For example, the earliest networks, those of Buffon in 1755 and Duchesne in 1766, illustrated within-species relationships in dog breeds and strawberry cultivars, respectively, so that contemporary taxa appeared at internal nodes (ie. some breeds or cultivars were seen as ancestors of others).
Lamarck's famous tree of 1809 showed relationships between higher taxonomic groups rather than species, and had several such groups transforming into other groups, so that the interior nodes represented contemporary taxonomic groups. His view of evolution was thus fundamentally different to that of Darwin.
Most of the subsequent pre-1859 trees of biological relationships showed non-genealogical affinity, for example those of Agassiz, Augier, Bronn, and Hitchcock — these were not intended to be evolutionary diagrams, because their authors did not believe in evolution (Ragan 2009; Tassy 2011). Other people followed the lead of Lamarck, and thus drew similar trees, such as Barbançois, Strickland, and Wallace.
Atkinson and Gray (2005) point out that "Darwinian ideas of descent with modification were less revolutionary in linguistics than they were in biology", and so Darwinian trees appeared earlier in linguistics. For example, Schlegel (1808) is usually credited with introducing a "stammbaum" (family tree) approach to comparative grammar, along with Bopp (1816). The previous language tree of Gallet in c.1800, showed a combination of geographical and chronological relationships, rather than being strictly genealogical, and some contemporary languages were shown at internal nodes. Both Čelakovský and Schleicher in 1853 independently drew the first truly genealogical diagrams in linguistics. These had contemporary languages at the leaves but language groups on the internal edges, rather than ancestral languages.
This leaves open the question of who first drew a tree that could be considered to be completely Darwinian.
The first tree
A family-tree approach has also been developed for textual analysis, where genealogical diagrams are called "stemma", and this is where we actually find the first diagrams that match all of Darwin's ideas about evolution, as listed above.
In 1827 Hans Samuel Collin and Carl Johan Schlyter published the first volume of the Corpus Iuris Sueo-Gotorum Antiqui, which was a compilation of all of the Medieval laws of Sweden, presented in both Latin and Swedish. Collin was involved as editor of volumes 1 and 2, with Schlyter acting as sole editor of volumes 3-13 (the latter published in 1877, so that Schlyter spent 55 years on the project).
In order to compile the definitive version of the laws, the editors consulted all of the known documents (some 800 or so), which consist of hand-written manuscript copies, each one being a copy of some earlier copy. The editors performed a detailed comparative analysis of the texts in order to establish an authemtic version of the original laws (their subsequent commentary in the books is longer than the laws themselves). This is, literally, a study of "descent with modification", and not merely an analogy with Darwin's famous expression.
The first volume, which covers the county laws of Västergötland, is unique among the 13 volumes in that the editors make the following comment in their Preface (page XXXVII):
Latin:
Quo evidentius appareat mutua illorum codicum nunc descriptorum ratio, qui continent textum Iuris VG. antiquioris vel recentioris, vel partem aliquam illius textus, hanc rationem, prout ex iis, in quibus inter se conveniunt aut differunt codices, iudicare potuimus, schemate quodam cognationis, Tab. III, exprimere tentavimus.
Swedish:
För att göra förhållandet emellan de nu beskrifna codices, som innehålla WGL:s text eller någon del deraf, enligt dess äldre eller yngre redaktion, så mycket mer åskådligt, hafva vi sökt att genom ett slags stamtafla, Tab. III, framställa deras slägtskap så som vi af deras inbördes öfverensstämmelser eller olikheter tryckt oss kunna sluta dertill.
Holm (1972) translates the Swedish as:There are two online copies of the book, in Google Books, but neither of these displays Figure III correctly, as it is apparently a foldout. So, I have included it here.
To make the relationship all the clearer between the codexes now described, containing in whole or in part the text of the Västergötland Law in its older or younger redaction, we have attempted to present their affinities, as far as we could determine them from mutual agreements and differences, in a kind of family-tree in Table III.
 |
| The 1827 stemma from Collin & Schlyter. The manuscript texts are lettered. The vertical axis represents time, with the dashed lines indicating 25-year intervals from 1300 to 1500. |
O'Hara (1996) points out that the idea of establishing the most authentic version of a text by reconstructing its ancestry may have been part of an earlier monastic tradition, designed to elucidate the nature of the original scriptures. However, Collin and Schlyter appear to be the first to have done this in such a thorough manner, as most people did not bother to locate all of the extant texts (see Holm 1972). Moreover, their use of a genealogical diagram to illustrate their conclusions seems to be totally original (Timpanaro 2005; Robins 2007). The stemma matches all of the Darwinian criteria, and so it lays claim to being the first Darwinian tree. Its most obvious difference to Darwin's ideas is that it refers to individual objects rather than to groups such as species.
Holm (1972) attributes the figure (and the idea for it) to Schlyter alone, although there is nothing in the original text to support this assumption — all of the editorial comments are written in the plural. However, Frederiksen (2009) has revisited the background to the stemma, and she concludes that "there is every possibility" that Schlyter should be given the sole credit. She also points out that if the stemma is "regarded as a schema that draws up the principal lines [of descent] and disregards the contamination of the tradition it would seem to be almost accurate." In other words, the editors seem to have got it right the first time.
The issue of contamination is an important one, referring to the fact that many textual copies are actually compiled form multiple sources. Under these circumstances the stemma should be a reticulating network not a tree. Indeed, Holm (1972) attributes the absence of stemma in any of the other 16 volumes to concern by Schlyter about contamination, and therefore the actual usefulness of a tree. Nevertheless, several other people produced stemma of varying degrees of sophistication soon after 1827, including Carl Zumpt in 1831, Friedrich Ritschl in 1832, and Johan Madvig in 1833 (Holm 1972; O'Hara 1996; Timpanaro 2005). For this blog, it is worth pointing out that the stemma by Ritschl (1832) explicitly shows contamination, and is thus a reticulate network, the first of its kind in stemmatology.
 |
| Hilgendorf's 1866 phylogeny of fossil snails. The fossils are aligned horizontally with respect to their geological layers. |
One very interesting feature of the Collin & Schlyter figure is its clear resemblance to the fossil diagrams produced independently by Franz Hilgendorf and Albert Gaudry in 1866. Both of these people studied fossils in situ, so that they could see their distribution in the geological layers, and the fossil record was complete enough for them to construct evolutionary scenarios that connected the fossils together. They thus both produced evolutionary trees with the vertical axis explicitly representing time. The only real difference from the stemma is that in their diagrams time proceeds from bottom to top (as do the fossil layers in situ).
Finally, it is worth noting one very modern feature on the stemma. In only one case is a manuscript indicated as being a direct descendant of another. In all other cases the internal nodes are unlabeled, so that the known texts show sister-group relationships rather than direct ancestor-descendant relationships. If we do not have independent evidence that an observed text (or fossil) is a direct ancestor of another text (or fossil), then we should not indicate it as such in the evolutionary history.
References
Atkinson QD, Gray RD (2005) Curious parallels and curious connections — phylogenetic thinking in biology and historical linguistics. Systematic Biology 54: 513-526.
Bopp F (1816) Über das Conjugationssystem der Sanskritsprache, in Vergleichung mit jenem der griechischen, lateinischen, persischen und germanischen Sprache. Andreäischen, Frankfurt-am-Main.
Collin HS, Schlyter CJ (eds) (1827) Corpus Iuris Sueo-Gotorum Antiqui. Volumen 1. Westgötalagen. Haeggström, Stockholm.
Frederiksen BO (2009) Stemmaet fra 1827 over Västgötalagen: en videnskabshistorisk bedrift og dens mulige forudsætninger. Arkiv för Nordisk Filologi 124: 129-150.
Holm G (1972) Carl Johan Schlyter and textual scholarship. Saga och Sed (Kungl. Gustav Adolfs Akademiens Årsbok 1972): 48-80.
Lamarck J-B (1809) Philosophie Zoologique. Dentu et l'Auteur, Paris.
O'Hara R (1996) Trees of history in systematics and philology. Memorie della Società Italiana di Scienze Naturali e del Museo Civico di Storia Naturale di Milano 27: 81-88.
Ragan MA (2009) Trees and networks before and after Darwin. Biology Direct 4: 43.
Ritschl F (1832) Thomae Magistri sive Theoduli Monachi Ecloga vocum Atticarum. Orphanotrophei, Halle.
Robins W (2007) Editing and evolution. Literature Compass 4: 89–120.
Schlegel F (1808) Über die Sprache und Weisheit der Indier: ein Beitrag zur Begrundung der Alterthumskunde. Mohr und Zimmer, Heidelberg.
Tassy, P. (2011) Trees before and after Darwin. Journal of Zoological Systematics and Evolutionary Research 49: 89-101.
Timpanaro S (2005) The Genesis of Lachmann's Method [translation]. University of Chicago Press, Chicago.
Wednesday, June 19, 2013
Using phylogenetic analyses for textual analysis
I have written before about the distinction between phylogenetic networks and other types of biological network (see Biological versus phylogenetic networks). Basically, a phylogenetic network starts with observed data and infers the network connections via some optimization procedure, whereas for most other networks the connections are the observed data and the network is summarized by one or more statistics such as Degree Centrality or Betweenness Centrality (for an explanation, see Network measures and phylogenetic networks).
This distinction is also important for the use of networks as data displays, both in biology and elsewhere. I have noted that splits networks, for example, are a very useful alternative to multivariate data-display analyses such as Principal Components Analysis (see Networks can outperform PCA ordinations in phylogenetic analysis). PCA can, for example, produce mathematical artifacts that distort the display, which is obviously undesirable (see Distortions and artifacts in Principal Components Analysis analysis of genome data).
It is interesting, therefore, to compare the different network types in terms of their ability to analyze and display a particular data set. To demonstrate the generality of the methods, here I discuss an analysis of a text document.
The one I have chosen has previously been analyzed by Seth Long (Text Network and Corpus Analysis of the Unabomber Manifesto). The Unabomber Manifesto is a 35,000 word document from 1995 entitled "Industrial Society and its Future", written by Theodore (Ted) Kaczynski, which is basically a critique of contemporary techno-capitalist society. A textual analysis is of interest because, as Seth notes: "The motives of all authors — or at least their traces — are always left behind in the lexical choices of their texts. Deliberate, written language is like a rhetorical fingerprint."
Textual analyses
Seth Long's textual analysis procedure was:
- import the text into an analytical tool (in this case AutoMap) in order to remove trivial words (eg. articles, conjunctions, pronouns), and to reduce inflected words to their base form;
- use the same tool to quantify what words are connected to what other words and how often (in this case using a two-word gap);
- import the result into a a network analysis tool (in this case Gephi) in order to visualize the semantic connections; each word is visualized as a node in the network, and words that appear next to each other appear as edges in the network.
The two most important network visualizations, in my opinion, show nodes with the highest levels of Betweenness Centrality and the highest levels of Degree Centrality. The latter measures how many total connections a node has to other individual nodes. The former measures whether or not a node is connected to other nodes that themselves have many connections.
In a textual network, a word with high degree centrality is a word used in connection with a myriad of other words. This simply tells you that a word is used frequently in a text and in a variety of contexts. A word with high betweenness centrality is a word used frequently and in conjunction with other words that also connect to other nodes to form community clusters. This tells you that a word is not only used frequently and not only in many contexts but also that it is used in connection with words that also do a lot of semantic work in the text. A word with high betweenness centrality is a word through which many meanings in a text circulate.
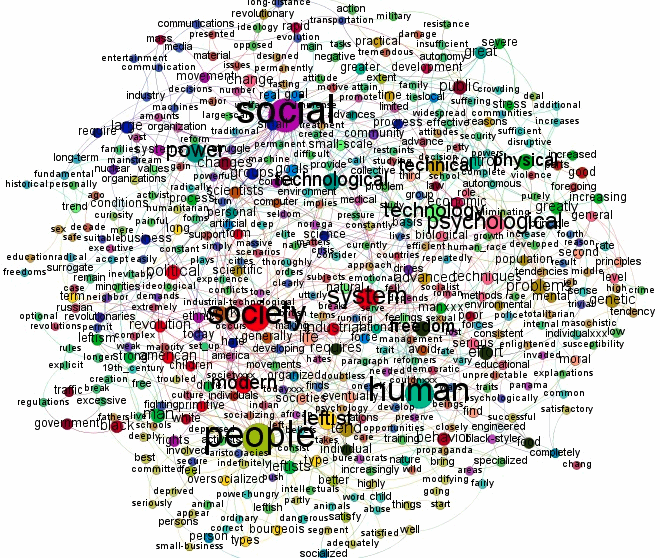 |
| Nodes with the greatest Degree Centrality in the text |
 |
| Nodes with the greatest Betweenness Centrality in the text |
The size of the words in the two networks represents their "amount" of centrality (ie. their importance). Clearly, these networks are very complex, and it would be best to simplify them. Seth does this in some of his other textual analyses, where he uses "one of Gephi’s degree range tools to hide the most disconnected nodes, thereby ‘cleaning’ the visualization of all but the most prominent clusters and connections" (eg. see Meaning circulation in Lolita). This has not been done in this example.
I will not provide an interpretation of these two networks, which you can find in Seth's original post. The basic conclusion is that "Kaczynski is a primitivist who loves nature more than humanity."
Seth also notes:
One thing a text network does, beyond providing an interesting visualization, is to point the researcher in the direction of terms and n-grams that might be explored more granularly in a corpus analysis tool, such as the NLTK [Natural Language Toolkit]. It provides a map of a text’s semantic circulation, a map that can be followed when we return to the world of pure textuality.The two corpus analyses that Seth Long provides are a histogram of the most frequent words, and a graph of where in the text the most frequent words fall (beginning, middle, end, throughout, etc).
Phylogenetic analyses
We can now compare these analyses to the use of phylogenetic trees and networks as heuristic tools for data analysis and display. The objective is the same as for the above analyses, and the general approach is also very similar. The main difference, as explained above, is that the nodes and edges are inferred rather than observed. This means that the words appear only at the ends of terminal edges, rather than being scattered throughout the network.
These analyses involve:
- remove trivial words, count the frequency of the remaining words, simplify the network by choosing how many of the words to display (50 in this case), and record their location in the text;
- calculate the semantic "distance" between words based on their co-occurrence in a sliding window (in this case 20 words), using some similarity measure (in this case the jaccard coefficient);
- visualize the distances as an unrooted phylogenetic tree, in this case a neighbor-joining tree calculated using TreeCloud;
- visualize the distances as an unrooted phylogenetic network, in this case a neighbor-net (a splits graph) calculated using SplitsNetworkCloud.
 |
| TreeCloud of the text |
 |
| NetworkCloud of the text |
In these phylogenetic analyses, the size of the words represents their frequency in the text, and the colour of the words represents their location in the text (red near the beginning, blue near the end). This adds the corpus analyses to the network visualization, making the graphs more informative. This can happen because the visualization itself is the inferred network, rather than the visualization summarizing various aspects of centrality of the underlying observed network.
The relative distance between two words in the text is given by the relative length of the path between them in the tree or network. Note that the "clean-up" of the graphs, by restricting the number of included words, helps a lot with the interpretation (as it would if also applied to the previous two graphs).
The phylogenetic tree focuses on certain of the word connections, rather than trying to display them all — it tries to infer which connections are "important" based on the measure of semantic distance, rather then connecting all nearby words. The interpretations from the tree are similar to those from the previous networks, but in many ways the interpretations are displayed more directly by the inferred (phylogenetic) graph than by summarizing centrality (either degree or betweenness).
Finally, the network is much more complex than the tree, which is often the case. Note that for the tree the edge lengths are all equal, but in the network the "average" distance between two words is given by the length of the path between them. This is just for illustrative purposes, as the tree could be drawn with variable edge lengths or the network drawn with unit edge lengths. The main reason for using unit edge lengths is that the terminal edges are often very long and the structure of the tree or network is hidden in the centre, as discussed by Gambette et al. (2012).
The main patterns that the network adds to the tree are: (a) "human" is separately associated with "control" and "behavior" on one hand and "beings" on the other; (b) "power" is separately associated with "process" and "autonomy" on one hand and "satisfy" on the other; and (c) "primitive" is separately associated with "individuals", "societies" and "groups" on one hand and "modern" and "man" on the other.
Conclusion
The phylogenetic approach is helpful because it focuses on certain of the network connections, rather than trying to display them all, as do the other networks. It cannot separately analyze concepts like degree and betweenness, and so information is lost; but this is traded off against the ability to include corpus analyses such as word frequency and location. Phylogenetic trees and networks can thus be valuable tools for textual analysis.
The TreeCloud was introduced by Gambette and Véronis (2010). If you read French, then examples are presented by Amstutz and Gambette (2010) and by Gambette and Martinez (2012) (the latter has a comparison with some other multivariate data anlayses).
Thanks to Philippe Gambette for producing the NetworkCloud.
References
Amstutz D., Gambette P. (2010) Utilisation de la visualisation en nuage arboré pour l'analyse littéraire. JADT'10: 10th International Conference on Statistical Analysis of Textual Data.
Gambette P., Gala N., Nasr A. (2012) Longueur de branches et arbres de mots. Corpus 11: 129-146.
Gambette P., Martinez W. (2012) L'affaire du Mediator au prisme de la textométrie. Manuscript.
Gambette P., Véronis J. (2010) Visualising a text with a tree cloud. In: Locarek-Junge H., Weihs C. (eds) Classification as a Tool of Research, Proceedings of IFCS'09 (11th Conference of the International Federation of Classification Societies), pp. 561-570.
Monday, June 17, 2013
Synapomorphies as tree slices
A phylogenetic tree is usually drawn as a stick diagram showing the historical lines of descent; and if the tree has been constructed from phenotypic data then the characters are often plotted onto the tree as cross-bars at the place where they are postulated to have arisen.
This creates an artistic problem if the phylogenetic tree is to be drawn as a real tree. Rob DeSalle & David Lindley (1997. The Science of Jurassic Park and the Lost World; or, How to Build a Dinosaur. Basic Books, ISBN 0-465-07379-4) have dealt with this issue by representing the synapomorphies as slices taken from the tree branches, as shown below.

Not all of the synapomorphies are shown, of course (birds have several more, and mammals do have some), but this is an intriguing artistic solution to a problem that Ernst Haeckel never conceived of when he first drew a phylogeny as a real tree. It is certainly more interesting than the more conventional representation (by Steven M. Carr), as shown below. (Note that mammals do acquire a synapomorphy in this version of the tree.)

Update note from Rob DeSalle:
"The figure was drawn by one of our artists here at the museum [American Museum of Natural History] and it was a collaborative effort to get the drawing done. In the conversation with the artist when outlining what I wanted in the figure I explained synapomorphy to him, and we went back and forth for an hour and settled on the slices. If I remember right it was the artist's idea. But the idea of representing synapomorphy as slices might have appeared before that."
Wednesday, June 12, 2013
Cophylogenetic networks
I want to talk about relationships between phylogenetic networks, with respect to the cophylogeny problem. This is a problem in which phylogenetic histories, which are typically trees, share some ecological link, which may be quite strict or relatively weak, such that their evolutionary dynamics are not independent. Given that nothing evolves in isolation, that there are more parasites than non-parasites, and that there are many genes – each of which can have its own phylogenetic history – for each organism that houses them, this is a ubiquitous scenario and one worthy of analysis.
A simple case serves to illustrate here:
 The tree on the left is labelled H for the "host" phylogeny but it could equally well be named the "species" phylogeny; the tree on the right is labelled P for the "parasite" or "pathogen" phylogeny but could also be thought of as the "gene" phylogeny. Reconciling these two, given the observed associations at the tips, is a computationally horrible problem (Libeskind-Hadas & Charleston 2010, Ovadia et al. 2011) already. The proof Ran Libeskind-and I did (he mostly did!) first allowed the host phylogeny to be a network but the later proof by Ovadia et al. didn't. But it's an interesting problem, because there are hybridization events of host species though not so much of individual genes.
The tree on the left is labelled H for the "host" phylogeny but it could equally well be named the "species" phylogeny; the tree on the right is labelled P for the "parasite" or "pathogen" phylogeny but could also be thought of as the "gene" phylogeny. Reconciling these two, given the observed associations at the tips, is a computationally horrible problem (Libeskind-Hadas & Charleston 2010, Ovadia et al. 2011) already. The proof Ran Libeskind-and I did (he mostly did!) first allowed the host phylogeny to be a network but the later proof by Ovadia et al. didn't. But it's an interesting problem, because there are hybridization events of host species though not so much of individual genes.
The cophylogeny problem is best attacked as a mapping problem (yes, this is my opinion: there are others). Given P, H and leaf associations phi, what is the minimal cost mapping of P into H that preserves phi and the structure of P and H, and which is interpretable in a well-defined way? (see Cophylogeny blog for more details.)
We typically have four event types:
We could in principle extend this to deal with failure-to-codiverge events:

Mike Steel once very usefully asked me, what are the desired properties of cophylogeny maps? In trying my best to answer him I realised that none of the properties really needs either phylogeny to be a tree. Nodes in P are mapped to nodes or edges in H, and the evolutionary history they imply is based on the route through H from parent node p' to child node p in P. If H is a tree then this is unique; otherwise there can be ambiguity and a potential explosion in number of solutions. But it does mean that potentially we can solve the mapping problem moderately well so long as P and H are at least DAGs. While this is possible in principle, in practice, it's pretty much impossible.
It's hard enough just with trees:
The figure above was cut from the manuscript and relegated to supplementary material because it was kind of unnecessary, but it's very pretty so it should see the light of day. It shows a consensus of 15 solutions / maps we found that could best explain the relationships between hosts and circoviruses; thicker lines for more frequently occurring components and different colours for different groups of maps.
The cophylogeny problem is a fun one that presents lots of modelling, computational and representational challenges.
A simple case serves to illustrate here:

The cophylogeny problem is best attacked as a mapping problem (yes, this is my opinion: there are others). Given P, H and leaf associations phi, what is the minimal cost mapping of P into H that preserves phi and the structure of P and H, and which is interpretable in a well-defined way? (see Cophylogeny blog for more details.)
We typically have four event types:
 |
| codivergence : a generalisation of cospeciation, where a node in P bifurcates at the same moment as does its host node in H; |
 |
| duplication : where the parasite bifurcates without a corresponding host bifurcation, such as for a gene duplication; |
 |
| host switch : a parasite/pathogen establishes on a new host lineage); and |
 |
| loss : we fail to see a parasite/pathogen where we expected it, caused by "missing the boat" / lineage sorting... |
 |
| extinction (above) or sampling failure. |

... but these cause new headaches.
Mike Steel once very usefully asked me, what are the desired properties of cophylogeny maps? In trying my best to answer him I realised that none of the properties really needs either phylogeny to be a tree. Nodes in P are mapped to nodes or edges in H, and the evolutionary history they imply is based on the route through H from parent node p' to child node p in P. If H is a tree then this is unique; otherwise there can be ambiguity and a potential explosion in number of solutions. But it does mean that potentially we can solve the mapping problem moderately well so long as P and H are at least DAGs. While this is possible in principle, in practice, it's pretty much impossible.
It's hard enough just with trees:
 |
| Figure from Ramsden et al. 2009 Supp. material |
The figure above was cut from the manuscript and relegated to supplementary material because it was kind of unnecessary, but it's very pretty so it should see the light of day. It shows a consensus of 15 solutions / maps we found that could best explain the relationships between hosts and circoviruses; thicker lines for more frequently occurring components and different colours for different groups of maps.
The cophylogeny problem is a fun one that presents lots of modelling, computational and representational challenges.
Monday, June 10, 2013
A network analysis of the Bundesliga
The German Fußball-Bundesliga association football (soccer) competition has just completed its 50th season, having started in 1963. This seems like an appropriate time to celebrate by performing a network analysis of the annual competition results. (I have previously provided a similar analysis for the results of the FIFA World Cup.)
The 1.Budesliga (the top level of the competition, with 18 teams) is one of the most prestigious in the world, and is currently ranked third in Europe (behind La Liga in Spain and the Premier League in England). In the current Elo Football Club Rankings, 15 of the top 250 ranked teams play in the Bundesliga, including the top-ranked team, FC Bayern München. (England and Spain both have 18 teams in the top 250, and Serie A in Italy has 16.)
Average attendance at the games is c. 45,000 people, with the average being >80,000 per game for the Borussia Dortmund team in the 2012-13 season. (This implies a stadium-capacity attendance at every Dortmund home game!) This national average is second only to the US National Football League (c. 65,000 people per game), and is far more than for any other soccer nation.
Of course, German national football competitions existed before the founding of the Bundesliga, but football was previously organized as five separate Oberligen (premier leagues) representing different geographical regions. The first Bundesliga competition involved 16 teams selected from these regions, starting in August 1963. The number of teams was expanded to 18 in 1965, where it has remained, except for the 1991-1992 season when there were 20 teams due to German reunification. A total of 52 clubs have competed in the 1.Bundesliga since it was founded, although only one team, Hamburger SV, has competed at the top level in all 50 seasons.
The relationships between these 52 teams represent a network within each competition, based on their relative success at the games they played (and thus their final position in the league table), and this network changes through time across the various seasons. It is this network of relationships across the years that I am analyzing here, using a phylogenetic network.
In this case, the phylogenetic analysis is being used in an exploratory manner — it is intended to be a convenient visual summary of the data. It seeks to uncover the patterns associated with a group of objects (teams in this case) for which multi-variable data have been collected (ie. competition results for multiple years). The network analysis assumes that the data have been formed by some historical process(es), and it produces a visualization that places objects with similar histories near each other in the network.
The analysis
I have taken the competition data for each season from the Deutsche Telekom T Online Sport page. The teams are simply ranked according to their official finishing position in each season. I have converted this to a score per season (1st=18points, 2nd=17, ... 18th=1point), with a score of zero if the team was absent from the 1.Bundesliga for that season. All 52 teams thus have a score each season from 0-18.
The similarity among the 50 scores for each pair of teams was calculated using the Steinhaus dissimilarity. The Steinhaus dissimilarity ignores "negative matches", as discussed in a previous blog post, so that two teams are not considered to have similar histories just because they were both absent from the 1.Bundesliga in the same years (ie. when they are absent their history is "unknown").
A Neighbor-net analysis was then used to display the between-team similarities as a phylogenetic network. This decomposes the similarities into a series of bi-partitions of the teams, and then tries to display as many of these bi-partitions as possible in two dimensions. Each bi-partition represents the division of the teams into two sub-groups, where the data indicate that the two sub-groups differ in some way. (See the post on How to interpret splits graphs.) That is, teams that are closely connected in the network are similar to each other based on their Bundesliga results, and those that are further apart are progressively more different from each other.

I have colour-coded some of the teams in order to highlight some of the patterns shown in the network:
| Brown | Those teams that have appeared in most of the seasons (43-50 out of 50), and have finished in the top 3 at least once |
| Pink | Teams finishing in the top 6 (ie. top third) at least once, and participated in at least 20 seasons (20-34) |
| Blue | Teams finishing in the top 8 at least once, and participated in 10-20 seasons |
| Purple | Teams finishing in the top 8 at least once, and participated in <10 seasons |
| Black | Teams rarely competing in the 1.Bundesliga, and not very successful when they have been there |
Note that most of these colours are strongly clustered in the network graph, especially the brown and pink ones. This is to be expected, because these teams have a very similar Bundesliga history.
The pink teams form two main clusters in the graph. These teams have been in the 1.Bundesliga for about half of the 50 seasons, and the two clusters represent which years they were present. That is, roughly speaking, when the five pink teams at the top of the graph were present then the four pink teams at the bottom were not, and vice versa. Bayer 04 Leverkusen is separate from the other pink teams because, rather than appearing in the 1.Bundesliga "on and off", it has been there continuously since 1979.
The blue teams form three groups in the graph. KFC Uerdingen 05 is separated from the other blue-coloured teams because its period of success was from 1975-1995, whereas the successes of Hansa Rostock, SC Freiburg and VfL Wolfsburg all date from 1993 onwards, and the successful years of DSC Arminia Bielefeld have been scattered throughout the 50 seasons.
The positions of the black and purple teams are based on similar considerations — their positions reflect which few seasons they participated in the 1.Bundesliga.
Note also that the very long terminal branches for most of the teams indicates that they actually do not have a great deal in common regarding their competition results. Teams have been successful to different extents in different years, relatively independently of each other. This is not surprising.
The largest split in the network separates the brown (10) and pink (10) teams from the others (plus Arminia Bielefeld), thus highlighting these 21 teams as the most successful competitors. In the network graph, the brown-coloured teams form a distinct subset of this group (along with Bayer 04 Leverkusen), forming the upper echelon of success.
With one exception (Vfl Wolfsburg), the competition champions have also come from this upper group — the number of championships is indicated in the network next to each of the 12 teams who have won. This highlights the unfortunate fact that both FC Schalke 04 (45 seasons, best finish 2nd) and Eintracht Frankfurt (43 seasons, best finish 3rd) have had consistent success without ever becoming champions.
This phylogenetic network thus provides a very effective visual summary of the main features of the 1.Bundesliga results when averaged over all of the 50 seasons.
Wednesday, June 5, 2013
Words lead to confusion in phylogenetics
In 2008 Hanno Sandvik, from the University of Tromsø in Norway, made an interesting observation about word confusion and the history of phylogenetics:
In passing I would like to mention a speculation on the reason why phylogenetic systematics and cladistic methodology was rather quickly accepted in Germany (Ax 1977; Remane 1956; Schlee 1969), but provoked intense debates in English-language journals (verifiable with almost any issue of Systematic Zoology from the 1970s). I suspect that part of the problem was semantic. The German word for ‘relationship’ is "Verwandtschaft", but while the English word has all kind of abstract and symbolic connotations, including overall similarity, the German term is reserved for true, genealogical bonds (as is the Norwegian "slekt"). The statement that for instance the lungfish is more closely "verwandt" to the cow than to the salmon is quite uncontroversial in German. On the other hand, the statement that the lungfish is more closely related to the cow than to the salmon, was able to create a heated discussion — which was only peripherally concerned with the actual phylogeny of the groups concerned (Gardiner et al. 1979; Halstead 1978; Halstead et al. 1979).This point seems to be directly relevant to the currently common uses of the expression "phylogenetic network", which can be seen to have an entirely homologous confusion. "Phylogenetic network" is used to describe both relationships based on general affinity (data-display or implicit networks) as well as the more restricted class of relationships based solely on genealogy (evolutionary or explicit networks). This duality potentially causes confusion every time the expression "phylogenetic network" is used, since it is not necessarily clear which meaning is intended. Fortunately, this has not (yet?!) lead to the sort of acrimonious "debates" that have been described for the introduction of cladistics to English-speaking biologists (Hull 1990; Felsenstein 2001).
This potential confusion also existed with the original introduction of what we would now call phylogenies, as this was done by French speakers — Buffon referred to his 1755 diagram as "une espèce d'arbre généalogique" [a type of genealogical tree], and Antoine Duchesne explicitly labelled his 1766 diagram as a "Généalogie". Similarly, Lamarck described his 1809 tree as "Servant à montrer l'origine des différens animaux" [Serving to show the origin of the different animals]. These are evolutionary diagrams, as their genealogical connotation is made clear. However, if this is translated into English as the generic word "relationship" then the clarity will disappear. [Tree-like diagrams produced by other authors at the same time usually referred to affinity, eg. "affinitates", "affinitatum".]
Incidentally, the word "phylogeny" is not now used with the original meaning given to it by Ernst Haeckel (Dayrat 2003). Haeckel used this word to describe a transformation series of character states, representing the order in which morphological changes have occurred through evolutionary time. These transformation series seem to owe more to Lamarck than to Darwin, as far as evolutionary ideas are concerned, but from them the tree of relationships could be constructed. For the tree itself Haeckel used the German word "stammbaum", which also has a directly genealogical meaning (eg. pedigree or family tree).
Finally, given the terminology that I have used above, it is confusing the discover the existence of Genealogical Implicit Affinity Networks!
References
Ax P. (1977) Willi Hennig. 20.4.1913 bis 5.11.1976 [Nachruf]. Verhandlungen der Deutschen Zoologischen Gesellschaft 70: 346-347.
Dayrat B. (2003) The roots of phylogeny: how did Haeckel build his trees? Systematic Biology 52: 515-527.
Felsenstein J. (2001) The troubled growth of statistical phylogenetics. Systematic Biology 50: 465-467.
Gardiner B.G., Janvier P., Patterson C., Forey P.L., Greenwood P.H., Miles R.S., Jefferies R.P.S. (1979) The salmon, the lungfish and the cow: a reply. Nature 277: 175-176.
Halstead L.B. (1978) The cladistic revolution — can it make the grade? Nature 276: 759-760.
Halstead L.B., White E.I., MacIntyre G.T. (1979) The salmon, the lungfish and the cow: L.B. Halstead and colleagues reply. Nature 277: 176.
Hull D.L. (1990) Science as a Process: an Evolutionary Account of the Social and Conceptual Development of Science. University Of Chicago Press.
Remane A. (1956) Die Grundlagen des natürlichen Systems, der vergleichenden Anatomie und der Phylogenetik, 2nd ed. Akademische Verlagsgesellschaft, Leipzig.
Sandvik H. (2008) Tree thinking cannot taken for granted: challenges for teaching phylogenetics. Theory in the Biosciences 127: 45-51.
Schlee D. (1969) Hennig’s principle of phylogenetic systematics, an ‘intuitive, statistico-phenetic taxonomy’? Systematic Zoology 18: 127-134.
Monday, June 3, 2013
Plotting evolutionary divergence and time
Evolutionary studies have always been based in some way on the phenotype / genotype distance between organisms, usually taken as representing divergence or convergence through evolutionary time. In phylogenetics, this aspect of evolution is usually secondary to the discovery of pathways of historical descent, which are based on patterns of character distribution among organisms rather than on their degree of character divergence.
Nevertheless, evolutionary biologists have long wanted a diagram that links the phenotype / genotype distance between organisms with evolutionary time. One could thereby visualize the presumed historical patterns of phenotype / genotype divergence (and convergence). This should produce a diagram that looks something like a phylogenetic tree (or a coalescent tree), except that one axis would explicitly represent time and the other would explicitly represent phenotype / genotype distance.
Mike Keesey, at the consistently interesting blog A Three-Pound Monkey Brain, has attempted to produce such a thing. The particular example shown here is based on All Known Great Ape Individuals (Messinian to Present).
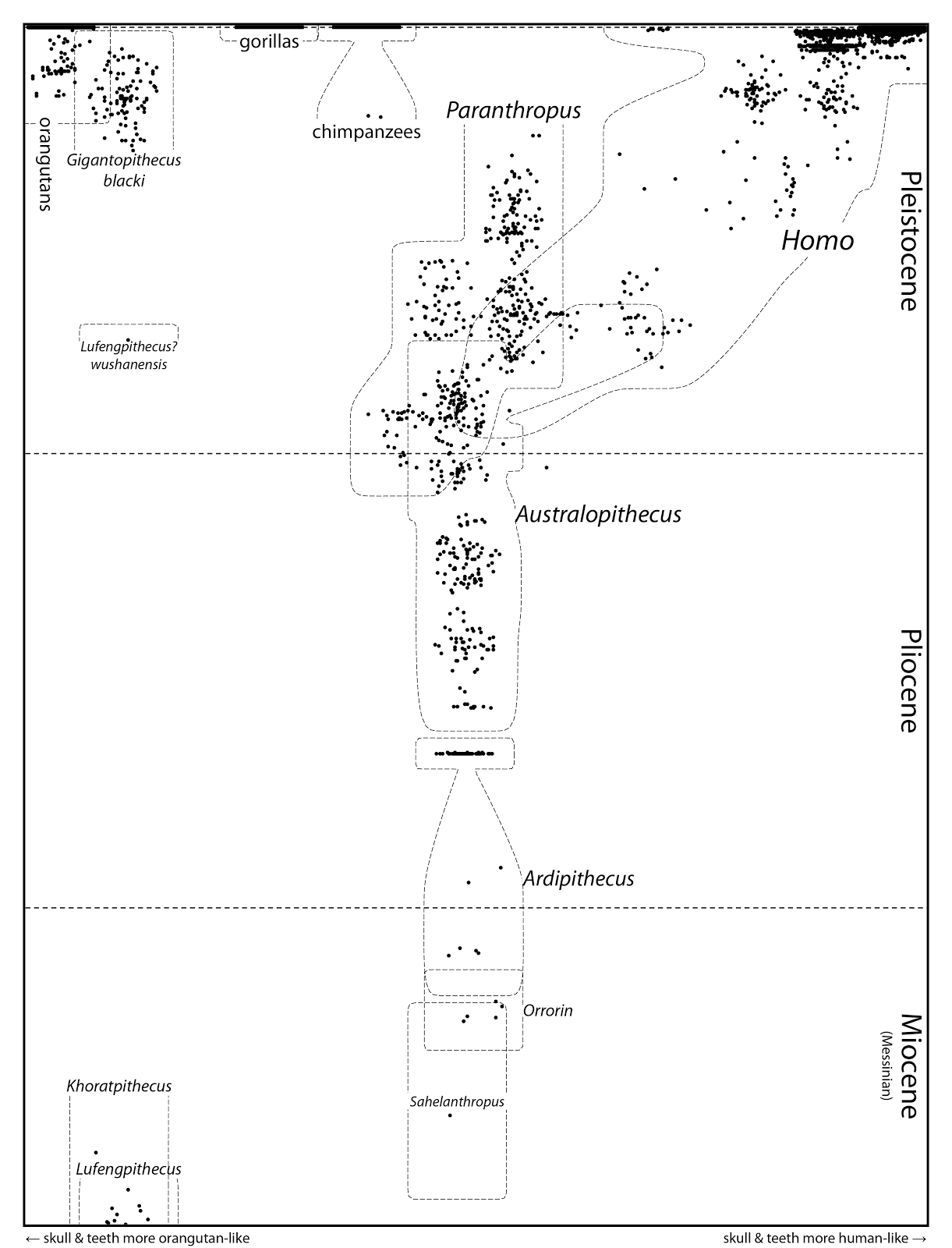
The figure is intended to include all known hominid fossil individuals (Hominidae), with the horizontal axis representing a distance matrix based on craniodental characters — varying from orangutan-like skull and teeth on the left to human-like on the right. Since the vertical axis represents fossil age, the diagram shows the morphological divergence of human-like individuals (including Neandertals) from earlier forms.
The diagram itself is somewhat of a work in progress, and Mike notes several limitations of the data (eg. absence of fossil gorillas and Pliocene stem-orangutans, lack of postcranial characters, the problematic assignment of names) as well as the analysis (eg. there's a random element to the plotting). Moreover, summarizing morphological divergence in only one axis is a major simplification, and so the diagram should not be over-interpreted.
Nevertheless, this looks very promising for those people who are interested in visualizing the process of evolutionary divergence. This could be especially useful if data are available for genetic distances rather than phenotypic ones. With the increasing availability of genome data for hominids, for example, the temporal relationships among Humans, Neandertals and Denisovans should be much easier to visualize (see Why do we still use trees for the Neandertal genealogy?). If nothing else, ancestral polymorphism would be clearly displayed!