I noted in an
earlier post that studies of the dog genealogy seem to follow historical precedent, with trees being used for the analysis of whole-genome data and networks for the analysis of mitochondrial DNA data. However, domestic dog breeds do not have a simple tree-like ancestry, due to the cross-breeding involved in creating new breeds, and so the use of a tree model is inadequate. This was known long before the advent of molecular data, from comparative studies of phenotypes rather than genotypes, but genetic data have allowed us to attack this issue in a more directly quantitative way.
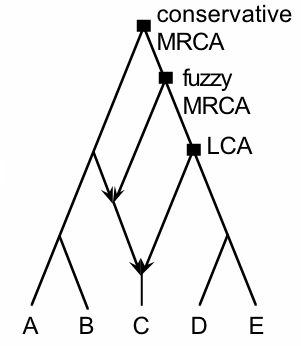
Anthropologists have traditionally used phylogenetic trees, especially when assessing the historical development of human "races", which have been assumed to maintain a strong degree of separation (see this
earlier post). Clearly, networks would be more appropriate representations of history in many cases, especially where there is gene flow within a species or set of closely related species. This particularly applies to those fossils most closely related to humans, such as those of the Neandertals, a group of archaic hominins from the Middle Pleistocene who ranged right across Europe into western Siberia, but whose fossil record stops about 30,000 years ago (during the Late Pleistocene).
There have been a number of recent blog comments about the desirability of network analyses in historical anthropology (e.g.
Dalton Luther,
Jonathan Marks,
PZ Myers,
Dienekes Pontikos). As noted by
Jason Antrosio, there "is a need to better understand and portray evolutionary complexity. With all the reports of Neandertal and Denisovan admixture, with all the emphasis on multispecies ethnography, with new looks at hybridization, we really must get away from the overly simplistic tree diagrams and taxonomies that have so long dominated evolutionary imagery". (Denisovans consist of a hominin fossil finger bone and some teeth from the Denisova Cave, in Siberia, which have yielded nucleotide sequences strikingly different from those of both Neandertals and modern humans. As
Todd Wood has noted: "they're a genome in search of a fossil record.")
Here, I use networks to evaluate some of the available genotype data for the relationships between humans, Neandertals and Denisovans.
Nuclear genome
There is currently very little whole-genome data for ancient hominins, but what there is clearly shows "that Neandertals, Denisovans, and others labelled archaic are in fact an interbreeding part of the modern human lineage ... There has been extensive admixture across modern humans for tens of thousands of years, and at least some admixture across several archaic groups" (from
Jason Antrosio again). Clearly, this is a situation for which networks were especially designed.
The relevant published papers include those on Denisovans (Reich et al. 2010, Meyer et al. 2012), Neandertals (Noonan et al. 2006, Green et al. 2006, 2010), an ancient human (Rasmussen et al. 2010), and the historical peopling of South-East Asia, Oceania and Australasia (Rasmussen et al. 2011, Reich et al. 2011, Skoglund and Jakobsson 2011, Mendez et al. 2012). Of these, only Mendez et al. and Meyer et al. used a network to analyze the evolutionary history (a Median-Joining network and an Admixture graph, respectively); the others used trees, ordinations and/or 3- and 4-taxon comparisons of genetic distances. The obvious question to ask is whether a tree is appropriate here.
As an example, we can take the "pairwise autosomal DNA sequence divergences" provided by Reich et al. (2010) for five of the genomes for which they collected SNP data. We cannot derive an evolutionary network directly from these data, but a data-display network will allow us to assess how tree-like are the data presented. Figure 1 shows a NeighborNet analysis of the data. This indicates that the data are strongly tree-like, mainly because of the authors concerted attempts to "clean up" the data from sequencing and analysis artifacts that would otherwise obscure the tree signal in ancient DNA. Nevertheless, there are still two detectable non-tree signals: one linking the Denisovan to the Neandertal from Mezmaiskaya (both fossil locations are in Russia), and a larger one linking the Denisovan to the Yoruba human (from a West African ethnic group). The first signal may represent non-tree gene flow, although the second signal is harder to explain (ancestral polymorphism, perhaps?).
 |
Figure 1. NeighborNet analysis of the autosomal DNA sequence
divergences for two modern humans (San, Yoruba), two fossil
Neandertals (Mezmaiskaya, Vindija), and a fossil Denisovan. |
Mitochondrial genome
Mitochondrial DNA (mtDNA) is the most commonly collected source of genetic data, especially sequences of the so-called control region (including the D-loop). Moreover, it is now quite commonplace to sequence the >16,500 nt of the mtDNA genome, as indicated by the contents of the
mtDB (Ingman and Gyllensten 2006) and
MitoTool (Fan and Yao 2011) databases. Mitochondrial DNA has also been successfully extracted from ancient hominins. Indeed, there are now sequences for the entire mtDNA genome of Denisovans (Krause et al. 2010a), Neandertals (Green et al. 2008, Briggs et al. 2009), and early modern humans (Ermini et al. 2008, Gilbert et al. 2008, Krause et al. 2010b). Compared to nuclear DNA, ancient mtDNA has a greater survival rate and greater degree of sequencing coverage, which leads to a markedly reduced influence of post-mortem damage and contamination (see Ho and Gilbert 2010).
The major assumed advantages of using mtDNA are (i) the high copy number, (ii) the maternal mode of inheritance, (iii) the high substitution rate (resulting in variation even at the intraspecific level), (iv) the lack of recombination (so that historical relationships can be modelled by a phylogenetic tree), and (v) the molecular clock is considered to be relatively reliable (so that the dates of historical events can be estimated). Both of these latter two assumptions have been disputed, however, as discussed by McVean (2001) for recombination and Endicott et al. (2009, 2010) for the clock.
The available data indicate that recombination in mtDNA is rare, if it occurs at all. Furthermore, gene flow is unlikely to complicate the historical relationships, because the mitochondrion is almost always inherited maternally and there is little evidence of historical movement by single females between populations, as opposed to movement by males. So, a phylogenetic tree is a reasonable model of evolutionary history for mtDNA, unlike the situation for the nuclear genome.
On the other hand, there are a number of issues that will make any attempt to reconstruct a tree problematic. That is, the data will not be tree-like, even if the genealogical history was tree-like. First, the genes in mtDNA are completely linked as a single locus, which will lead to deep coalescence (incomplete lineage sorting), thus disconnecting gene history and organism history. Second, mtDNA exhibits considerable heterogeneity in nucleotide-substitution rates along the genome, with the control region having very high rates (up to 10x that of the reset of the mtDNA) and codon second positions having very low rates. Indeed, it is likely that substitutional saturation occurs in the control region, and that purifying selection occurs at first and second codon positions. There will be an enormous amount of homoplasy under these circumstances (eg. parallel substitutions). Third, there is evidence of different nucleotide-substitution rates in different lineages, even when those lineages are closely related. This will also cause homoplasy.
There have been three responses to these problems by those who study human mtDNA. First, trimming of the sequence data occurs. For example, there are well-known nucleotide positions that are usually deleted because their variation seems random, and others whose excessive variation leads them to be down-weighted. Second, a network is used to assess how non-tree-like are the data. People have developed several network methods explicitly for mtDNA data, such as Median-Joining and Reduced-Median networks; and the literature is replete with papers using these methods to analyze mtDNA sequences. Third, a partitioned model is needed in order to build a phylogenetic tree. Notably, the different codon positions need separate substitution models, as do the control region and the RNA-coding regions. Furthermore, rate heterogeneity needs to be modelled, and a relaxed molecular clock is needed.
 |
Figure 2. An approximate Median Network (based on a
Median-Joining analysis) of control region sequences from
13 fossil Neandertals and 1 fossil Denisovan. |
These problems are bad enough for the study of within-human phylogenies, but they are even more problematic for the study of ancient DNA. For example, substitutional saturation means that the control region, and especially the three hypervariable regions (HVR1,HVR2,HVR3) that are the most frequently sequenced parts of it, is almost useless for reconstructing ancient history. This can be seen, for example, in the data of Dalén et al. (2012), who analyzed the mtDNA control regions of 13 Neandertals and 1 Denisovan. Dalén et al. produced a bayesian tree from these data, but in Figure 2 I show a Median Network instead. (This displays all of the maximum-parsimony trees simultaneously.) There may well be an evolutionary tree in these data, but if so then it is pretty deeply buried, and it is unlikely to be recovered reliably without a lot of work.
Unfortunately, for the study of ancient DNA very little seems to be done about the problems of homoplasy, in terms of any of the three suggested solutions. Indeed, most of the concern seems to be about potential post mortem damage to the DNA (eg. extra substitutions in the terminal branches), instead. For example, I have checked 21 empirical phylogenetic studies involving Neandertal mtDNA (published since 1997), and only 6 of them noted that they had either down-weighted or excluded particular hyper-variable nucleotide positions: Krings et al. (1999, 2000), Caramelli et al. (2006), Ermini et al. (2008), Moradi and Schuster (2008) and Endicott et al. (2010). Second, only three of the papers presented an empirical network analysis: Ermini et al. (2008) (a Reduced-Median network), Caramelli et al. (2006) (a Median-Joining network) and Caramelli et al. (2008) (a TCS network); for the rest, they either presented a tree, an ordination, or no empirical diagram at all. Third, only two of the analyses performed a partitioned tree-building analysis: Green et al. (2008) and Endicott et al. (2010). Finally, 14 of the 21 papers were based on sequences of the control region only, which makes their phylogenetic inferences questionable.
If I concentrate here on the production of a phylogenetic network, as I should be doing in this blog, then it is will become obvious why tree-building analyses are rather difficult for Neandertal sequence data. Figure 3 uses a data-display network to show the non-tree features of the available Neandertal mtDNA genomes. Note that there is very little common variation at all, meaning that Neanderthal mtDNA has very limited genetic variation. Moreover, there are
no tree-like parts to the diagram, with every parsimony-informative nucleotide position being contradicted by at least one other. Analyzing these data with a simple tree-building analysis seems to be inappropriate, to say the least.
 |
Figure 3. Median Network analysis of the six full-length mtDNA
genomes currently available for Neandertals. The numbers on the
branches indicate the number of characters that change along
each branch. |
To assess the relationship between Neandertals and humans (which seems to be the most common ancient-DNA question addressed in the literature), we can add the Denisovan mtDNA sequence, plus the 3 available sequences for early modern humans, and also some sequences from a range of modern humans (ie. the revised Cambridge Reference Sequence, plus 53 sequences from Ingman et al. 2000). However, we then cannot plot the Median Network because several of the aligned positions are no longer binary (ie. they are not SNPs). So, I will use a NeighborNet analysis for the data display instead, as shown in Figure 4. The first thing to note is that the genetic variation in the Neanderthal mtDNA is much less than that in the human mtDNA, and probably less than can be accounted for solely by the smaller sample size (6 genomes versus 54).
 |
Figure 4. NeighborNet analysis of the mtDNA genomes from
6 Neandertals, 1 Denisovan, 3 early modern humans and 54
contemporary humans, based on uncorrected genetic distances. |
Second, there is clearly an underlying tree-like structure to the data, as expected, which I have emphasized by plotting the related Neighbor-Joining tree for comparison in Figure 5 (the NeighborNet analysis is a generalization of the Neighbor-Joining tree). However, there is just as clearly considerable non-tree structure to the data, notably in the relationship of the Denisovan sequence to the other sequences, but also in the relationship between the Neandertals and the humans. It is this non-tree structure that complicates any attempt to reconstruct the evolutionary relationship of the Neandertals to humans; and it appears to result, at least partly, from the homoplasy caused by saturation of nucleotide substitutions.
 |
| Figure 5. Neighbor-Joining tree of the same data used for Figure 4. |
However, even the NeighborNet analysis cannot summarize
all of the non-tree patterns in the data, but presents instead a selective summary of them. To get further insight into the extent of the problem, I have deleted the 53 human sequences, and then plotted the Pruned Quasi-median network in Figure 6. This network is the equivalent of the Median Network while allowing for non-binary sequence positions. It is difficult to believe that these data were created by a simple tree-like evolutionary process, and, if so, that it will be easy to reconstruct it.
 |
Figure 6. Pruned Quasi-median network analysis of the mtDNA
genomes from 6 Neandertals, 1 Denisovan, 3 early modern humans
and 1 contemporary human (the revised Cambridge Reference
Sequence). The branch lengths are not drawn to scale. |
Anyway, the most-common network approach to trying to untangle this sort of mess in mtDNA sequence data is to use either a Reduced-Median network or a Median-Joining network, which are simplifications of the full Median Network. I have produced a Median-Joining network in Figure 7, as an example. The interesting thing to note here is that the Denisovan sequence does not connect to the rest of the network between the Neandertal cluster and the human cluster of sequences, which it does do in all of the published phylogenetic trees. This pattern is not unexpected, given the pattern shown in the Pruned Quasi-median network (Figure 6), but it does suggest that the tree-building analyses performed to date are somewhat naïve in the face of considerable sequence complexity, by not explicitly dealing with that complexity.
 |
Figure 7. Median-Joining network analysis of the same data used
for Figure 4. Only the sequences from Figure 6 are labelled — the
other dots are the remaining 53 contemporary humans, plus some
inferred ancestors. The branch lengths are not drawn to scale. |
Conclusion
The phylogenetic analysis of Neandertal mtDNA has been critiqued a number of times before (eg. Gutiérrez et al. 2002, Hebsgaard et al. 2007, Moradi and Schuster 2008, Endicott et al. 2010). However, this has always been in the context of "providing a better tree-building analysis", rather than in the context of evaluating and displaying the conflicting information that complicates the tree-building analysis, as I have done here. In this context, it is important to note that none of the diagrams that I have produced here are evolutionary networks, and so they do not represent a reconstruction of evolutionary history. They are intended merely to display the convoluted nature of the ancient mtDNA sequence data, and to emphasize the valuable role that phylogenetic networks can play in evaluating such data.
One further point worth noting is that these diagrams are all unrooted, which neatly avoids the problems associated with adding a chimpanzee sequence in order to locate the root of the evolutionary history. Adding this sequence dramatically increases the sequence complexity, of course. In particular, the nuclear genome apparently places the Denisovan as the sister to the Neandertals whereas the mtDNA places it as the sister to Neandertals+humans (eg. note that the mid-point rooting of Figure 5 would be on the branch leading to the Denisovan).
References
Briggs A.W., Good J.M., Green R.E., Krause J., Maricic T., Stenzel U., Lalueza-Fox C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Schmitz R., Doronichev V.B., Golovanova L.V., de la Rasilla M., Fortea J., Rosas A., Paabo S. (2009) Targeted retrieval and analysis of five Neandertal mtDNA genomes.
Science 325: 318-321.
Caramelli D., Lalueza-Fox C., Condemi S., Longo L., Milani L., Manfredini A., de Saint Pierre M., Adoni F., Lari M., Giunti P., Ricci S., Casoli A., Calafell F., Mallegni F., Bertranpetit J., Stanyon R., Bertorelle G., Barbujani G. (2006) A highly divergent mtDNA sequence in a Neandertal individual from Italy.
Current Biology 16: R630-R632.
Caramelli D., Milani L., Vai S., Modi A., Peccholi E., Girardi M., Pilli E., Lari M., Lippi B., Ronchitelli A., Mallegni F., Casoli A., Bertorelle G., Barbujani G. (2008) A 28,000 years old Cro-Magnon mtDNA sequence differs from all potentially contaminating modern sequences.
PLoS One 3: e2700.
Dalén L., Orlando L., Shapiro B., Durling M.B., Quam R., Gilbert T.M.P., Díez Fernández-Lomana C.J., Willerslev E., Arsuaga J.L., Götherström A. (2012) Partial genetic turnover in neandertals: continuity in the east and population replacement in the west.
Molecular Biology and Evolution 29: 1893-1897.
Endicott P., Ho S.Y.W., Metspalu M., Stringer C. (2009) Evaluating the mitochondrial timescale of human evolution.
Trends in Ecology and Evolution 24: 515-521.
Endicott P., Ho S.Y.W., Stringer C. (2010) Using genetic evidence to evaluate four palaeoanthropological hypotheses for the timing of Neanderthal and modern human origins.
Journal of Human Evolution 59: 87-95.
Ermini L., Olivieri C., Rizzi E., Corti G., Bonnal R., Soares P., Luciani S., Marota I., De Bellis G., Richards M.B., Rollo F. (2008) Complete mitochondrial genome sequence of the Tyrolean Iceman.
Current Biology 18: 1687-1693.
Fan L., Yao Y.G. (2011) MitoTool: a web server for the analysis and retrieval of human mitochondrial DNA sequence variations.
Mitochondrion 11: 351-356.
Gilbert M.T., Kivisild T., Grønnow B., Andersen P.K., Metspalu E., Reidla M., Tamm E., Axelsson E., Götherström A., Campos P.F., Rasmussen M., Metspalu M., Higham T.F., Schwenninger J.L., Nathan R., De Hoog C.J., Koch A., Møller L.N., Andreasen C., Meldgaard M., Villems R., Bendixen C., Willerslev E. (2008) Paleo-Eskimo mtDNA genome reveals matrilineal discontinuity in Greenland.
Science 320: 1787-1789.
Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H., Hansen N.F., Durand E.Y., Malaspinas A.S., Jensen J.D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H.A., Good J.M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E.S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Doronichev V.B., Golovanova L.V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R.W., Johnson P.L., Eichler E.E., Falush D., Birney E., Mullikin J.C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S. (2010) A draft sequence of the Neandertal genome.
Science 328: 710-722.
Green R.E., Krause J., Ptak S.E., Briggs A.W., Ronan M.T., Simons J.F., Du L., Egholm M., Rothberg J.M., Paunovic M., Pääbo S. (2006) Analysis of one million base pairs of Neanderthal DNA.
Nature 444: 330-336.
Green R.E., Malaspinas A.S., Krause J., Briggs A.W., Johnson P.L., Uhler C., Meyer M., Good J.M., Maricic T., Stenzel U., Prüfer K., Siebauer M., Burbano H.A., Ronan M., Rothberg J.M., Egholm M., Rudan P., Brajković D., Kućan Z., Gusić I., Wikström M., Laakkonen L., Kelso J., Slatkin M., Pääbo S. (2008) A complete neandertal mitochondrial genome sequence determined by high-throughput sequencing.
Cell 134: 416-426.
Gutiérrez G., Sánchez D., Marín A. (2002) A reanalysis of the ancient mitochondrial DNA sequences recovered from Neandertal bones.
Molecular Biology and Evolution 19: 1359-1366.
Hebsgaard M.B., Wiuf C., Gilbert M.T., Glenner H., Willerslev E. (2007) Evaluating Neanderthal genetics and phylogeny.
Journal of Molecular Evolution 64:50-60.
Ho S.Y.W., Gilbert M.T.P. (2010) Ancient mitogenomics.
Mitochondrion 10: 1-11.
Ingman M., Gyllensten U. (2006) mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences.
Nucleic Acids Research 34: D749–D751.
Ingman M., Kaessmann H., Pääbo S., Gyllensten U. (2000) Mitochondrial genome variation and the origin of modern humans.
Nature 408: 708-713.
Krause J., Briggs A.W., Kircher M., Maricic T., Zwyns N., Derevianko A., Pääbo S. (2010b) A complete mtDNA genome of an early modern human from Kostenki, Russia.
Current Biology 20: 231-236.
Krause J., Fu Q., Good J.M., Viola B., Shunkov M.V., Derevianko A.P., Paabo S. (2010a) The complete mitochondrial DNA genome of an unknown hominin from southern Siberia.
Nature 464: 894-897.
Krings M., Geisert H., Schmitz R.W., Krainitzki H., Pääbo S. (1999) DNA sequence of the mitochondrial hypervariable region II from the Neandertal type specimen.
Proceedings of the National Academy of Sciences of the USA 96: 5581-5585.
Krings M., Capelli C., Tschentscher F., Geisert H., Meyer S., von Haeseler A., Grossschmidt K., Possnert G., Paunovic M., Pääbo S. (2000) A view of Neandertal genetic diversity.
Nature Genetics 26: 144-146.
McVean G.A.T. (2001) What do patterns of genetic variability reveal about mitochondrial recombination?
Heredity 87: 613-620.
Mendez F.L., Watkins J.C., Hammer M.F. (2012) Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations.
Molecular Biology and Evolution 29: 1513-1520.
Meyer M., Kircher M., Gansauge M.-T., Li H., Racimo F., Mallick S., Schraiber J.G., Jay F., Prüfer K., de Filippo C., Sudmant P.H., Alkan C., Fu Q., Do R., Rohland N., Tandon A., Siebauer M., Green R.E., Bryc K., Briggs A.W., Stenzel U., Dabney J., Shendure J., Kitzman J., Hammer M.F., Shunkov M.V., Derevianko A.P., Patterson N., Andrés A.M., Eichler E.E., Slatkin M., Reich D., Kelso J., Pääbo S. (2012) A high-coverage genome sequence from an archaic Denisovan individual.
Science (advance)
Moradi C.R., Schuster A. (2008) Evaluation of the critical factors in the phylogenetic analysis of human and neanderthal mtDNA.
Unpublished ms.
Noonan J.P., Coop G., Kudaravalli S., Smith D., Krause J., Alessi J., Chen F., Platt D., Pääbo S., Pritchard J.K., Rubin E.M. (2006) Sequencing and analysis of Neanderthal genomic DNA.
Science 314: 1113-1118.
Rasmussen M., Guo X., Wang Y., Lohmueller K.E., Rasmussen S., Albrechtsen A., Skotte L., Lindgreen S., Metspalu M., Jombart T., Kivisild T., Zhai W., Eriksson A., Manica A., Orlando L., De La Vega F.M., Tridico S., Metspalu E., Nielsen K., Ávila-Arcos M.C., Moreno-Mayar J.V., Muller C., Dortch J., Gilbert M.T., Lund O., Wesolowska A., Karmin M., Weinert L.A., Wang B., Li J., Tai S., Xiao F., Hanihara T., van Driem G., Jha A.R., Ricaut F.X., de Knijff P., Migliano A.B., Gallego Romero I., Kristiansen K., Lambert D.M., Brunak S., Forster P., Brinkmann B., Nehlich O., Bunce M., Richards M., Gupta R., Bustamante C.D., Krogh A., Foley R.A., Lahr M.M., Balloux F., Sicheritz-Pontén T., Villems R., Nielsen R., Wang J., Willerslev E. (2011) An Aboriginal Australian genome reveals separate human dispersals into Asia.
Science 334: 94-98.
Rasmussen M., Li Y., Lindgreen S., Pedersen J.S., Albrechtsen A., Moltke I., Metspalu M., Metspalu E., Kivisild T., Gupta R., Bertalan M., Nielsen K., Gilbert M.T., Wang Y., Raghavan M., Campos P.F., Kamp H.M., Wilson A.S., Gledhill A., Tridico S., Bunce M., Lorenzen E.D., Binladen J., Guo X., Zhao J., Zhang X., Zhang H., Li Z., Chen M., Orlando L., Kristiansen K., Bak M., Tommerup N., Bendixen C., Pierre T.L., Grønnow B., Meldgaard M., Andreasen C., Fedorova S.A., Osipova L.P., Higham T.F., Ramsey C.B., Hansen T.V., Nielsen F.C., Crawford M.H., Brunak S., Sicheritz-Pontén T., Villems R., Nielsen R., Krogh A., Wang J., Willerslev E. (2010) Ancient human genome sequence of an extinct Palaeo-Eskimo.
Nature 463: 757-762.
Reich D., Green R.E., Kircher M., Krause J., Patterson N., Durand E.Y., Viola B., Briggs A.W., Stenzel U., Johnson P.L.F. (2010) Genetic history of an archaic hominin group from Denisova Cave in Siberia.
Nature 468: 1053-1060.
Reich D., Patterson N., Kircher M., Delfin F., Nandineni M.R., Pugach I., Ko A.M., Ko Y.-C., Jinam T.A., Phipps M.E., Saitou N., Wollstein A., Kayser M., Pääbo S., Stoneking M. (2011) Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania.
American Journal of Human Genetics 89: 516-528.
Skoglund P., Jakobsson M. (2011) Archaic human ancestry in East Asia.
Proceedings of the National Academy of Sciences of the USA 108: 18301-18306.